
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCombined electrochemical and microscopic study of porous enzymatic electrodes with direct electron transfer mechanism†
M.
Varničić
a,
K.
Bettenbrock
a,
D.
Hermsdorf
a,
T.
Vidaković-Koch
*a and
K.
Sundmacher
ab
aMax Planck Institute for Dynamics of Complex Technical Systems, Sandtorstraße 1, 39106 Magdeburg, Germany. E-mail: vidakovic@mpi-magdeburg.mpg.de
bOtto-von-Guericke University Magdeburg, Universitätsplatz 2, 39106 Magdeburg, Germany
First published on 4th August 2014
Abstract
In the present work electrochemical and microscopic methods have been utilized to get more insight into the complex relationship between the preparation route, structure and activity of porous enzymatic electrodes. Enzymatic electrodes have been prepared following two procedures. In one procedure enzymes were physically entrapped into a porous conductive matrix stabilized by “inert” binder (Vulcan-PVDF), while in the second one (Vulcan-Gelatin) gelatin has been used as a binder and the electrodes were cross-linked. Vulcan-PVDF electrodes show exceptionally high activity (up to 1.2 mA cm−2) compared to Vulcan-Gelatin electrodes (0.3 mA cm−2) at nominally lower enzyme loading. The scanning electron microscopy cross-sections of these electrodes revealed similar thicknesses, but a higher level of Vulcan nanomaterial agglomeration, somewhat reduced porosity and formation of gelatin film on top in the case of Vulcan-Gelatin electrodes. Additionally, fluorescence microscopy studies provided evidence of a higher level of enzyme agglomeration in the case of cross-linking. Although the gelatin matrix and the reduced catalyst layer porosity might slow down hydrogen peroxide diffusion, Vulcan-Gelatin electrodes are less affected by mass transfer conditions than Vulcan-PVDF electrodes. A plausible cause of the Vulcan-Gelatin electrode inferior performance is a lower number of active enzymes (lower enzyme utilization) compared to the Vulcan-PVDF electrode caused by a higher level of enzyme agglomeration in former case.
1. Introduction
Broader applications of redox enzymes as catalysts in bio-based technical systems like enzymatic fuel cells, bio-batteries or bioelectrochemical reactors require significant increase of the catalytic current per geometrical surface area of the electrode. This goal can be possibly achieved by improvement of the electrode structure, for example by introduction of high surface area materials, resulting in 3-D electrodes.1–4 3-D structuring introduces various materials into electrode design; enzymes as catalytic elements, additives like hydrogels for enhancing the enzyme stability and various nanomaterials as conductive supports for immobilization of the biocatalyst. In addition, suitable mediators might be required if the enzyme does not allow direct electron transfer (DET). All these components are commonly self-organized in the catalyst layer and their dispersion is unknown. A similar problem has been faced in the field of conventional gas diffusion electrodes, where the designer task is to create a large so-called 3-phase interface. In case of enzymatic electrodes and DET, for optimal design, enzymes should be contacted by both an electron- and ion-conductive phases such that the reaction can take place. It can be easily anticipated that the formation of enzyme agglomerates, which might result from some preparation procedures, will drastically reduce the enzyme utilization. Similarly, additional components in the catalyst layer, like different hydrogels might break the electron conductive network, rendering parts of the catalyst layer inactive. It clearly follows that understanding of the relationship between the preparation conditions and the electrode performance is crucial for the optimal design of enzymatic electrodes.Experimental methods for preparation of enzymatic electrodes can be roughly classified into two groups. First group of methods is based on physical immobilization of enzymes. The simplest approach is physical adsorption where only weak interactions between a support and an enzyme are involved. As supports, electron conductive materials like gold or carbon surfaces or in the case of 3-D electrodes, different types of nanomaterials have been typically used.5–8 It has been demonstrated that using this method high performance enzymatic electrodes can be prepared even without any surface modification in order to promote DET.8 Another possibility for physical immobilization is entrapment of enzymes into gel matrixes such as gelatin, collagen and polysaccharides. This approach usually stabilizes enzymes more than only physical adsorption.9–11 Second group of methods is based on chemical immobilization of enzymes. These methods include covalent enzyme immobilization on the electrode surface which requires functionalization of supports to create surface chemical groups for enzyme binding. Various surface modifications have been described in literature providing carboxyl, epoxy, acetyl or amino groups. These surface groups can be further either directly linked to enzymes or by using additional cross linkers like glutaraldehyde.12
Although methods based on covalent attachment have major benefit of higher enzyme stability at the expanse of somewhat lower activity due to reduction in enzyme flexibility13 and in some cases oriented enzyme immobilization can be achieved proving especially beneficial in case of the DET,14 we concentrate in the present paper on physical methods for enzyme immobilization. The major goal is to check how the preparation procedure influences electrode structural parameters like porosity and the electrode thickness. A further question is how enzyme organization at the conductive surface is dependent on the preparation procedure. To answer these questions porous enzymatic electrodes following two main routes of physical enzyme immobilization i.e. physical adsorption into porous structure and enzyme entrapment into gelatin matrix stabilized by cross-linking have been prepared. As a model enzyme horseradish peroxidase showing DET has been chosen. These electrodes have been characterized electrochemically for hydrogen peroxide reduction. Several factors which can influence electrode activity like: electrode surface area, thickness, enzyme distribution and agglomeration have been hypothesized. To prove their influence on observed activity, the electrodes, in addition to electrochemical methods, have been characterized using different microscopic methods. Scanning electron microscopy (SEM) has been used in order to get information on overall electrode structure (porosity, and its thickness), while fluorescence microscopy has been employed to visualize enzyme distribution on different supports.
2. Experimental part
2.1 Reagents
Horseradish peroxidase (EC 1.11.1.7, HRP) from Amorica rusticana was supplied from Serva Electrophoresis GmbH. Hydrogen peroxide (H2O2, 30 wt%) and gelatin were purchased from Merck. The H2O2 solution (3%) was prepared daily by dilution of 30% hydrogen peroxide. Poly(vinylidene fluoride) (PVDF), glutaraldehyde (GA) and 1-methyle-2-pyrrolidone were supplied by Sigma Aldrich. For fluorescence measurements DyLight 350 NHS ester dye, supplied by Thermo scientific with an excitation wavelength of 353 nm and an emission wavelength of 432 nm was used. All chemicals were of analytical reagent grade and all solutions were prepared using ultrapure water from Millipore.2.2 Preparation of enzyme modified surfaces
For electrochemical measurements spectroscopically pure carbon (SPG) rods with impurities equal to or less than 2 ppm supplied by Ted Pella, 330 INC, USA were cut in 11 mm diameter discs and have been used as supports for enzyme modification. Before modification, they were polished by fine emery paper (P1000), rinsed with deionized water and then further polished with ordinary white paper to smoothen the surface.15,16 For preparation of HRP modified electrodes, 50 μl of HRP solution in phosphate buffer (6 mg ml−1, pH 6.00) was placed on the top of the SPG disc and left for 2 h under ambient conditions. After that it was washed with distillated water and used for measurements. The discs have been mounted in a sample holder for rotating disc electrode experiments (RDE, Radiometer Analytical, model ED101) with an opening of 6 mm.Cross-linked electrodes were prepared by dipping enzyme modified discs in GA solution (5% in water) for 1 min, rinsing with water and drying at room temperature.17
Porous enzymatic electrodes incorporating enzymes and carbon nanoparticles (Vulcan XC72R supplied by Cabot Corporation) have been prepared by following two different procedures. In the first procedure, denoted in text as “Vulcan-Gelatin”, gelatin has been used as a binder and electrodes were cross-linked. This procedure was similar to procedure reported by Ivanov et al.18 Briefly, 20 mg of carbon nanomaterial and 10 mg HRP were suspended in 2% gelatin at 37 °C and cast on stainless steel discs degreased with acetone before usage. Electrodes were subsequently dried at ambient temperature and afterwards cross linked as previously described.
In the second procedure denoted in the text as “Vulcan-PVDF”, poly(vinylidene fluoride) (PVDF) was used as a binder material. This procedure was similar to those described by Tsujimura et al.7 Shortly, carbon nanomaterial was dissolved in 0.25 wt% PVDF solution in 1-methyle-2-pyrrolidone. In the next step, the ink was cast on SPG discs and left to dry at 60 °C. After drying, electrodes were ready for modification with HRP solution. Adsorption of enzyme on Vulcan electrodes was performed for 2 h at room temperature by applying 50 μl of 6 mg ml−1 HRP solution in 0.1 M phosphate buffer. Electrodes were then rinsed with buffer and were ready for use.
For atomic force microscopy (AFM) experiments highly oriented pyrolytic graphite (HOPG), supplied also from Ted Pella, 330 INC, USA was cut in the size of 5 mm × 4 mm. For preparation of HRP electrodes, a droplet of diluted HRP solution in phosphate buffer (6 mg ml−1, pH 6.00) was placed on the top of the HOPG and left to dry. The dilution was made in order to obtain a monolayer on the HOPG surface.
For fluorescence microscopy experiments, both SPG and HOPG supports have been used. Before surface modification, HRP was labeled in the following way: HRP solution (2 mg ml−1, pH 7.00) was mixed with fluorescence dye dissolved in dimethylformamide (DMF) and left for 1 h at room temperature. Afterwards, the excess non-reacted dye was removed by dialysis for 4 h using three dialysis buffer changes. The labeled enzymes were stored at 4 °C. For modification of SPG and HOPG surfaces, a droplet of diluted HRP solution was applied on an appropriate surface and left to dry.
2.3 Measurements
Electrochemical experiments were performed using Autolab potentiostat (PGSTAT302, Eco Chemie). Saturated calomel (SCE) and Pt electrodes were used as reference and counter electrodes, respectively. The electrolyte was a 0.1 M phosphate buffer with pH 6.0. All electrochemical experiments have been done under nitrogen atmosphere at 400 rpm (rounds per minute). Steady state polarization curves were obtained by extracting the current values after 60 s at constant potential values.Fluorescence microscopy has been performed with Imager M1 Microscope, Carl Zeiss. The objective was EC Plan Neofluar and filter set with excitation 365, beamer splitter 395 and emission 445/50 were used. In order to obtain high-contrast images and at the same time to avoid saturation, different exposure times have been used for different images (for further information please see the respective figure captions).
AFM measurements have been performed in air using 5500 SPM (Agilent Technologies), with tapping mode (Acoustic AC Mode). A rectangular silicon cantilever (PPP-NCSTAuD, Nanosensors) with a nominal force constant of 7.4 N m−1 has been used for the measurements.
Cross-sectional scanning electron microscopy (SEM) analysis of the enzymatic electrodes was performed using XL30 FEG (FEI Company).
3. Results and discussion
Porous enzymatic electrodes prepared based on two immobilization strategies described in the Experimental section have been tested for their activities towards hydrogen peroxide reduction (Fig. 1). Shortly, in one procedure, enzymes are immobilized by physical entrapment into a porous structure stabilized by “inert” binder (Vulcan-PVDF) while in the other one “active” binder (gelatin) and cross-linking to form and stabilize enzyme/nanoparticle composites (Vulcan-Gelatin) has been used. The performances of the enzymatic electrodes have been evaluated by means of cyclic voltammetry and steady state measurements. Fig. 1 shows cyclic voltammograms of the Vulcan-PVDF (Fig. 1a) and Vulcan-Gelatin (Fig. 1b) electrodes in phosphate buffer in absence and in presence of hydrogen peroxide, in quiet solution (0 rpm) and at 400 rpm rotation. As can be seen after addition of hydrogen peroxide, an increase of the reduction current can be observed indicating biocatalytic reduction of H2O2 by HRP. According to expectations16 the electrode activity in quiet solution is lower than at 400 rpm. This is especially true for Vulcan-PVDF electrode (current density (after background current subtraction) in the limiting current region is ca. 0.66 mA cm−2 with and 0.13 mA cm−2 without stirring), while Vulcan-Gelatin electrode is less affected by stirring conditions (current density in the limiting current region is ca. 0.22 mA cm−2 with and 0.13 mA cm−2 without stirring). These results indicate stronger mass transfer limitations in the Vulcan-PVDF compared to Vulcan-Gelatin case. Furthermore, these results suggest higher amount of active enzymes in the Vulcan-PVDF case.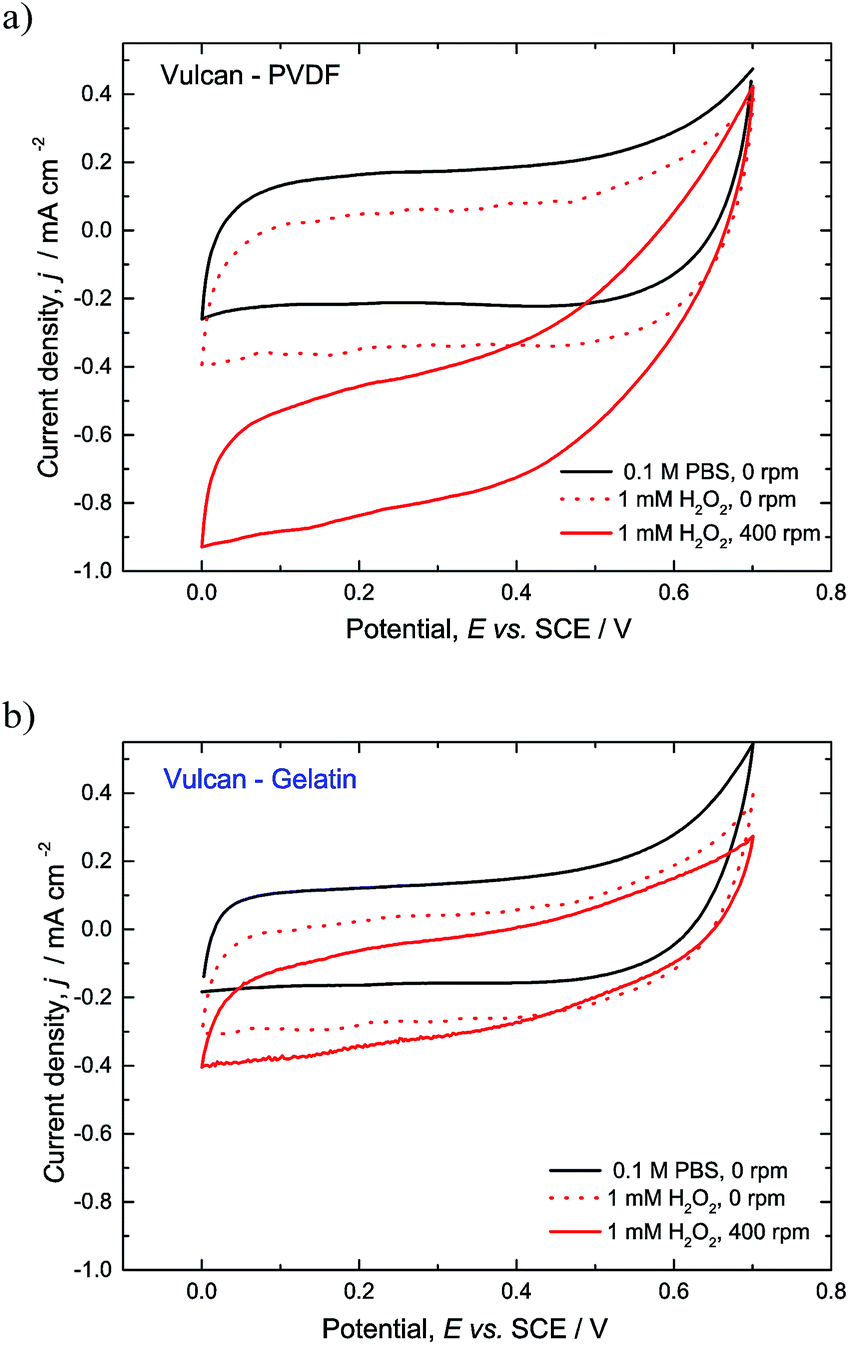 | ||
| Fig. 1 Cyclic voltammograms of porous enzymatic electrodes in phosphate buffer and in 1 mM hydrogen peroxide, without rotation and at 400 rpm. (a) Vulcan-PVDF and (b) Vulcan-Gelatin. Conditions: scan rate 5 mV s−1, pH 6.00, room temperature, N2 atmosphere, enzyme loadings (1 mg cm−2 for Vulcan-PVDF and 1.75 mg cm−2 for Vulcan-Gelatin electrodes). | ||
The activity of Vulcan-Gelatin electrode in terms of current densities is similar to reported values in literature, while Vulcan-PVDF electrode outperforms all literature results. Some examples are composite electrodes made of HRP immobilized on carbon nanotubes19 with activity of ca. 0.1 mA cm−2 at 1 mM hydrogen peroxide concentration in the limiting current region, or HRP immobilized on single walls carbon nanotubes (SWCNT) with activity of ca. 0.03 mA cm−2 at 0.3 mM hydrogen peroxide concentration.20 It should be stressed out that for fair comparison of electrode activities in different publications, some benchmarking is necessary. This benchmarking includes in addition to substrate concentration, pH and temperature, control of mass transfer resistance in the Nernstian diffusion layer and the same method for sampling of current–potential data. The control of mass transfer resistance in the diffusion layer over time can be only achieved under forced convection conditions. This control is very important for quantitative description of electrode processes since in quiet solution the thickness of the diffusion layer is changing over time of the experiment in a quantitatively unpredictable manner. This appears especially important in processes strongly controlled by mass transfer. For example in the present case the activity of Vulcan-PVDF electrode is highly underestimated under non-stirred conditions in comparison to Vulcan-Gelatin electrode.
The influence of the sampling method has been demonstrated in Fig. 2 below. The comparison between background subtracted cyclic voltammogram at 5 mV s−1 with steady state measurements at two different sampling times (60 and 120 s), show that the cyclic voltammetry overestimates significantly the catalytic current. Regarding the sampling time, one can see that the results after 60 s and 120 s are almost identical for which reason 60 s sampling time has been chosen in further measurements. The chronoaperometric data which have been used for construction of steady state current density–potential relationship are presented in Fig. S1 (ESI†).
 | ||
| Fig. 2 Comparison of the electrode performance obtained using cyclic voltammetry and steady state methods. (a) Vulcan-PVDF and (b) Vulcan-Gelatin electrodes. Conditions: scan rate 5 mV s−1 pH 6.00, N2 atmosphere, enzyme loadings: Vulcan-PVDF – 1 mg cm−2 and Vulcan-Gelatin – 1.75 mg cm−2. | ||
Having in mind previous discussion, forced convection conditions at constant rotation rate of 400 rpm and steady state method with sampling time of 60 s have been fixed in further measurements.
As can be seen in Fig. 2 both electrodes show high onset potential values (ca. 0.62 and 0.57 V vs. SCE for Vulcan-PVDF and Vulcan-Gelatin electrodes respectively), comparable with literature values on high surface area electrodes e.g. 0.57 V vs. SCE at pH 6,20 0.55 V vs. SCE at pH 7,19 and 0.63 V vs. SCE at pH 7.00.21 The onset potential value of the Vulcan-PVDF electrode, is ca. 50 mV more positive than the measured value for Vulcan-Gelatin electrode. In general, for the same type of peroxidase, onset potential values depend on pH of the solution,22 on peroxide concentration, (with more negative onset potential values at lower concentrations) and on immobilization procedure. The later effect might impact enzyme orientation at the surface as well as the number of active enzymes. It can be anticipated that both issues might contribute to observed differences between onset potentials of Vulcan-PVDF and Vulcan-Gelatin electrodes. In the case of Vulcan-PVDF electrodes enzymes were only physically adsorbed, while in the case of Vulcan-Gelatin procedure they were also cross-linked. One can hypothesize that enzyme cross-linking causes less favored enzyme orientations than the physical adsorption of enzymes (Vulcan-PVDF case) resulting in more negative onset potential. As it was discussed the results in Fig. 1 and 2 indicate higher number of active enzymes in the case of Vulcan-PVDF electrode. Alternatively at the same number of active enzymes, lowering of the kinetic constants of cross-linked enzymes could also explain the experimental observations. These two effects can not be separated, without being able to quantify the number of active enzymes (ref. 16 and references therein).
Next, the influence of enzyme loading at constant peroxide concentration has been checked for both immobilization procedures (Fig. 3). As can be seen in Fig. 2 Vulcan-PVDF electrodes are more active than Vulcan-Gelatin electrodes in the whole range of studied loadings. The dependences of current densities at constant potential (0.0 V vs. SCE) on enzyme loading show a bell-shaped form with optimal loading at ca. 1 mg cm−2 and 1.75 mg cm−2 for Vulcan-PVDF and Vulcan-Gelatin electrodes respectively. While an initial increase of the activity with enzyme loading can be correlated with an increase of the number of active enzymes, decrease of activity at higher enzyme loadings might be a consequence of a mass transfer resistance increase, in the catalyst layer at higher enzyme loadings.
 | ||
| Fig. 3 Influence of enzyme loading on the activity of Vulcan HRP – electrodes. Conditions: 1 mM hydrogen peroxide, electrode potential 0.0 V vs. SCE, 400 rpm, N2 atmosphere, pH 6.00. | ||
As a consequence dead portions of the catalyst layer can be created, which are under supplied with substrate.23 In addition to this reason unfavoured enzyme orientation is often commented in literature as a possible cause of activity decrease at higher loadings.21 Regarding different optimal loadings for two different procedures there are several reasons which can contribute to this observation. In accordance to our recent modeling study23 utilization of the catalyst layer depends on the thickness of the layer, its porosity, number of active enzymes and the concentration of reactant. In addition at higher hydrogen peroxide concentrations the effect of enzyme inhibition can become evident.24 The results indicate a lower number of active enzymes in Vulcan-Gelatin case. The reduced number of active enzymes, at the same concentration of reactant can cause better utilization of the catalyst layer in case of Vulcan-Gelatin electrode shifting position of the maximum to higher enzyme loadings.
The effect of hydrogen peroxide concentration was further studied for two optimal loadings of Vulcan-PVDF and Vulcan-Gelatin electrodes (Fig. 4). The increase of reduction current with an increase of hydrogen peroxide concentration indicates that the immobilized HRP retains its catalytic activity for the reduction of hydrogen peroxide. The results show that saturation conditions are reached at ca. 5 mM and ca. 4 mM hydrogen peroxide concentration for Vulcan-PVDF and Vulcan-Gelatin electrodes respectively. The Vulcan-PVDF electrode has excellent performance comparable with performance of bilirubin oxidase (BOD) based biocathode prepared on Ketjen Black (KB) suggesting that also these HRP-enzymatic electrodes are suitable for biofuel cell application.25
 | ||
| Fig. 4 Influence of the hydrogen peroxide concentration on the activity of Vulcan HRP – electrodes at optimized enzyme loadings. Conditions: enzyme loadings 1.75 mg cm−2 for Vulcan-Gelatin and 1.1 mg cm−2 for Vulcan-PVDF, electrode potential 0.0 V vs. SCE, 400 rpm, N2 atmosphere, pH 6.00. | ||
To understand the origin of the high activity of Vulcan-PVDF and lower activity of Vulcan-Gelatin electrodes, these two electrodes have been further characterized electrochemically in the absence of hydrogen peroxide as well as physically with SEM. The electrochemical characterization in the absence of active component (hydrogen peroxide) gives a rough orientation on active surface area available for enzyme adsorption. As can be seen in Fig. 5, the CVs of both electrodes in absence of hydrogen peroxide appear almost identical, showing only characteristic features of carbon material.26 Although Vulcan-Gelatin electrode had a bit higher Vulcan loading (3.6 mg cm−2) than Vulcan-PVDF electrode (3.0 mg cm−2), the results indicate similar active surface area for enzyme adsorption.
 | ||
| Fig. 5 Cyclic voltammograms of Vulcan HRP – electrodes in 0.1 M phosphate buffer. Conditions: scan rate 20 mV s−1, 400 rpm, N2 atmosphere, pH 6.00. | ||
Furthermore cross-sections of two types of electrodes have been studied by SEM (Fig. 6). It can be seen that the thicknesses of two electrodes are 47 μm and 53 μm for Vulcan-Gelatin and Vulcan-PVDF electrodes respectively. Magnification of the electrode cross sections (Fig. 6c and d) provides better insights into electrode morphology. Vulcan nanomaterial in Vulcan-Gelatin electrode forms knot-shaped agglomerates with the size around 250 nm which are significantly bigger than unit-structures in the Vulcan-PVDF electrode. This can be due to hydrophilic nature of gelatin, resulting in a higher degree of agglomeration of hydrophobic Vulcan nanoparticles. If PVDF was used, distribution of Vulcan nanoparticles is more uniform and ca. 100 nm spherical units can be observed (Fig. 6d). This indicate lessening of available surface area in the case of Vulcan-Gelatin electrode compared to Vulcan-PVDF electrode, which is also in accordance to electrochemical characterization (Fig. 5) where the CVs of both electrodes appear very similar despite a bit higher Vulcan loading of Vulcan-Gelatin electrode.
 | ||
| Fig. 6 SEM images of Vulcan HRP electrodes: cross sections of Vulcan-Gelatin (a) and Vulcan-PVDF (b); magnified view of the cross-sections of Vulcan-Gelatin (c) and Vulcan-PVDF (d) and top views of Vulcan-Gelatin (e) and Vulcan-PVDF (f). | ||
The top views of the Vulcan-Gelatin and Vulcan-PVDF electrode surfaces are also affected by preparation conditions as shown in Fig. 6e and f. Vulcan-Gelatin electrode has a layer of gelatin on the top which additionally stabilizes the electrode structure, might prevent/decrease leaching of enzymes, but introduces additional mass transfer resistance for hydrogen peroxide transfer in the catalyst layer. The surface of Vulcan-PVDF electrode has similar morphology to the electrode cross section. Additionally, porosities of both electrodes have been estimated based on the electrode thickness measured by SEM and theoretical compact electrode thickness based on loadings of all electrode components and their densities, according to the equation provided by Gode et al.27 Taking into account density of dry gelatin, the estimated value of electrode porosity for Vulcan-Gelatin procedure is 0.27. Calculated porosity for the Vulcan-PVDF electrode of 0.45 indicates lower mass transport limitations through the electrode layer in comparison to Vulcan-Gelatin electrodes.
The missing part of information is the influence of cross-linking on enzyme distribution and organization in the catalyst layer. Enzymes are protein structures having a size of several nm.28 They have been visualized by different microscopic methods like atomic force microscopy (AFM)29–33 scanning electrochemical microscopy (SECM), electrochemical scanning tunnelling microscopy (ESTM)34 and fluorescence microscopy (FM).35 In the present work FM has been applied for studying the enzyme organization on conductive supports. This method provides optical images of enzymes on surfaces, utilizing either their native fluorescence (e.g. flavin enzymes (FAD)36 are fluorescent) or more common foreign fluorescence obtained by labelling of enzymes with fluorescent markers. It has been so far successfully applied to investigate interactions of proteins entrapped in different polymers35,37–39 and for verification of enzyme self-assembly layer formation on the microarray electrodes.40,41
Since, HRP lacks its natural fluorescence it was first modified with amine-reactive dye containing N-hydroxysuccinimide (NHS) ester which is one of the most commonly used reactive groups for protein labeling. Modification occurs through formation of covalent bonds between the NHS ester and surface-oriented primary amines of the protein.42 In order to verify that the labeling procedure was successful, the electrophoresis of modified and unmodified enzymes has been done. Prepared gel contained two lanes, one with labeled HRP and one with unlabeled HRP. After separation and before applying standard staining procedure (in order to color all separated proteins), gel was observed under UV lamp (Fig. S2a ESI†). In this way, only fluorescent proteins are visualized. In the second step, after staining procedure, all proteins have been visualized. As can be seen in Fig. S2b ESI,† in both lanes HRP with molecular weight of 44 kDa was observed at the expected position and only the labeled protein showed fluorescence properties (Fig. S2a ESI†). It has been already shown that the presence of label does not perturb significantly the behavior of the enzymes.35 However, in order to verify that the labeling procedure does not affect the enzyme properties in the present case, the electrochemical activities of enzymatic electrodes modified with labeled and non-labeled enzymes were compared. The performances of these electrodes were found to be almost identical, indicating that the labeling did not affect the activity of the enzyme on the electrode surface (data not shown).
The following fluorescence microscopy measurements have been performed on model surfaces, but we believe that they provide good indications on enzyme distribution inside of porous structures. The effects of surface roughness and the cross-linking on enzyme distribution have been checked. To test the influence of the surface roughness, labeled HRP has been physically adsorbed on HOPG (ideally flat surface) and SPG (roughness factor 5 (ref. 43)). In both cases the quantity of an adsorbed enzyme corresponded to the calculated monolayer coverage. In case of the HRP–HOPG surface, the fluorescent image (Fig. 7a) shows a uniform level of fluorescence across the whole surface, which can be probably associated with a uniform distribution of enzymes on the flat HOPG surface. To check the flatness of the surface and the assumption of monolayer enzyme coverage, the HOPG surface before and after the modification was screened by AFM. The roughness of unmodified HOPG was found to be low with a maximum height of the profile below 0.4 nm (Fig. S3 ESI†). The AFM images with physically distributed enzyme patterns at relatively large areas (Fig. 7b). This pattern formation was strongly pH sensitive, showing for example more expressed branchy-like structures at pH 7.2 (Fig. S4 ESI†). The average heights of these structures are ca. 4 nm and ca. 5 nm at pH 6 and pH 7.2, respectively. These values correspond well to reported values of HRP dimensions (6.2 × 4.3 × 1.2 nm (ref. 3 and 34)) indicating monolayer formation. Interestingly, although the height of enzyme aggregates is not pH dependent, the width of these aggregates is pH sensitive (ca. 100 nm and ca. 300 nm at pH 6 and pH 7.2, respectively; Fig. 7b and S4 ESI†). This can possibly have an impact on the resulting enzyme catalytic activity.
 | ||
| Fig. 7 Labeled-HRP on HOPG support visualized by fluorescence microscopy with exposure time of 200 ms (a) and AFM image of HRP adsorbed on HOPG in a monolayer at pH 6 (b). | ||
Unlike the HRP–HOPG surface, the fluorescence image of HRP–graphite surface (Fig. 8a) shows a non-uniform distribution of fluorescence with blue spots of different intensities as well as very dark areas. These dark areas have a very low level of fluorescence (ca. 200 A.U.) and can be probably ascribed to enzyme-free parts of the surface. The blue spots with different intensities indicate a non-uniform distribution of enzymes on the remaining part of the surface, with spots showing a higher level of fluorescence (ca. 1500 A.U.) probably indicating enzyme agglomeration, while spots with a lower level of fluorescence (ca. 800 A.U.) (similar to those observed on HOPG surface) indicating monolayer enzyme adsorption.
 | ||
| Fig. 8 Fluorescence microscopy images of labeled-HRP on the SPG before rotation, 200 ms exposure time (a) and after 2 h rotation in RDE, 400 rpm, 100 ms exposure time (b). | ||
The non-uniform distribution of enzymes on the graphite surface corresponds well to the higher level of its surface inhomogeneity compared to HOPG. This result suggests that a monolayer of enzymes can be formed only on ideally flat surfaces like HOPG. If the roughness of the surface is of higher order than the size of the enzyme one can always expect the formation of agglomerates and consequently a non-uniform enzyme distribution. It can be further anticipated that the adsorption strength between the enzyme and the surface will vary for different adsorption sites like flat areas or depressions on the surface. This is confirmed by the image in Fig. 8b where the graphite surface after pronounced electrode rotation is shown. One can easily see that the blue spots of lower intensity, which were assigned to monolayer adsorption, disappeared. The enzyme distribution on the surface has a significant influence on the enzyme activity, especially in the case of DET, where the enzyme's active centers should be in close proximity of the electrode to allow for electron transfer. Our results indicate that in addition to orientation, enzyme agglomeration decreases the number of enzymes being in direct contact with the electrode surface.
The effect of cross-linking has been studied on HOPG and graphite surfaces (Fig. 9). According to literature cross-linking increases enzyme stability without influencing its activity (except in case of extremely high ratios between cross-linker and enzymes).44
 | ||
| Fig. 9 Fluorescence microscopy images of labeled-HRP after CL using 5% glutaraldehyde on HOPG, imaged with the exposure time of 100 ms (a) and on spectroscopic graphite surface with 200 ms exposure time (b). | ||
In addition, cross-linking is responsible for formation of enzyme agglomerates which can be clearly seen on both HOPG and spectroscopic graphite surfaces (Fig. 9a and b). While on HOPG one large agglomerate forms, on spectroscopic graphite “agglomeration centers” which differ in shape and size can be observed. The average level of fluorescence for these cross linked agglomerates on spectroscopic graphite is ca. 3200 ± 300 A.U. (profile shown only for one agglomerate), while the level of fluorescence for agglomerates on graphite without CL has values of ca. 1500 A.U. It can be anticipated that formation of enzyme-agglomerates decreases the number of active enzymes in contact with the electrode surface, which reduces further bio-electrode activity. This has been confirmed in experiment where the activities of non and cross-linked electrodes have been compared (Fig. 10). On the other hand CL increases the stability of the electrode, probably by decreasing the level of leaching. The calculated loss of activity after 2 hours at constant potential of 0.0 V vs. SCE was ca. 13% for CL electrode and ca. 28% for the non-cross-linked electrode (data not shown).
 | ||
| Fig. 10 Steady state polarization curves of hydrogen peroxide reduction on the HRP-modified graphite electrodes without and with cross linking. Conditions: 160 μM hydrogen peroxide concentration, 400 rpm, N2 atmosphere, pH 6.00. | ||
4. Conclusions
In this study porous enzymatic electrodes have been prepared by following immobilization protocols with and without cross-linking. It was demonstrated that the electrodes without hydrogels and further stabilization through cross-linking show significantly higher activities for the same nominal enzyme loading. Optimized HRP-enzymatic electrodes exhibit high activity towards hydrogen peroxide reduction reaching current density of ca. 1.2 mA cm−2, which according to our knowledge has not been reported in literature so far.The electrochemical characterization in the absence of reactant hydrogen peroxide has shown that both electrodes have almost the same electrochemically active surface area. SEM cross sections demonstrate that the thicknesses of two electrodes were similar, but porosity of Vulcan-Gelatin electrode was reduced in comparison to Vulcan-PVDF electrode. It was shown that addition of gelatin leads to stronger agglomeration of Vulcan nanomaterial. In addition gelatin forms a film on top, which can cause mass transfer limitations. The fluorescence microscopy studies on model surfaces have demonstrated that level of enzyme agglomeration depends on surface roughness and it increases upon cross-linking. This has a negative effect on electrode activity in both onset potential values and overall activity. Physical adsorption leads to uniform enzyme distribution only in the case of ideally flat surfaces. On macroscopically flat surfaces, enzyme agglomerates are also formed but in less extent compared to cross-linked conditions. The surface utilization for enzyme adsorption is very small.
Although the presence of gelatin matrix and the reduced porosity in Vulcan-Gelatin electrodes might slow down significantly mass transfer of the substrate through these electrodes, Vulcan-Gelatin electrodes are less affected by mass transfer conditions than Vulcan-PVDF electrodes. This implies higher reaction resistance in the case of Vulcan-Gelatin. According to our results, higher reaction resistance is caused by smaller number of active enzymes or by lowering of the kinetic constants of cross-linked enzymes. These two effects can not be separated, without being able to quantify the number of active enzymes.
Acknowledgements
The authors gratefully acknowledge the support of Helga Tietgens and Markus Ikert, during fluorescence microscopy analysis and AFM measurements and group of Dr Erdmann Rapp, for support with electrophoresis.Notes and references
- I. Ivanov, T. Vidakovic-Koch and K. Sundmacher, Energies, 2010, 3, 803–846 CrossRef CAS PubMed.
- T. Tamaki, Top. Catal., 2012, 55, 1162–1180 CrossRef CAS.
- M. H. Osman, A. A. Shah and F. C. Walsh, Biosens. Bioelectron., 2011, 26, 3087–3102 CrossRef CAS PubMed.
- J. A. Cracknell, K. A. Vincent and F. A. Armstrong, Chem. Rev., 2008, 108, 2439–2461 CrossRef CAS PubMed.
- X. J. Wang, M. Falk, R. Ortiz, H. Matsumura, J. Bobacka, R. Ludwig, M. Bergelin, L. Gorton and S. Shleev, Biosens. Bioelectron., 2012, 31, 219–225 CrossRef CAS PubMed.
- T. Kihara, X. Y. Liu, C. Nakamura, K. M. Park, S. W. Han, D. J. Qian, K. Kawasaki, N. A. Zorin, S. Yasuda, K. Hata, T. Wakayama and J. Miyake, Int. J. Hydrogen Energy, 2011, 36, 7523–7529 CrossRef CAS PubMed.
- S. Tsujimura, Y. Kamitaka and K. Kano, Fuel Cells, 2007, 7, 463–469 CrossRef CAS.
- D. Svedruzic, J. L. Blackburn, R. C. Tenent, J. D. R. Rocha, T. B. Vinzant, M. J. Heben and P. W. King, J. Am. Chem. Soc., 2011, 133, 4299–4306 CrossRef CAS PubMed.
- E. H. Yu and K. Scott, Energies, 2010, 3, 23–42 CrossRef CAS PubMed.
- J. Kim, H. F. Jia and P. Wang, Biotechnol. Adv., 2006, 24, 296–308 CrossRef CAS PubMed.
- E. T. Hwang and M. B. Gu, Eng. Life Sci., 2013, 13, 49–61 CrossRef CAS.
- C. F. Meunier, X. Y. Yang, J. C. Rooke and B. L. Su, ChemCatChem, 2011, 3, 476–488 CrossRef CAS.
- M. J. Cooney, V. Svoboda, C. Lau, G. Martin and S. D. Minteer, Energy Environ. Sci., 2008, 1, 320–337 CAS.
- O. Rudiger, C. Gutierrez-Sanchez, D. Olea, I. A. C. Pereira, M. Velez, V. M. Fernandez and A. L. De Lacey, Electroanalysis, 2010, 22, 776–783 CrossRef.
- R. Andreu, E. E. Ferapontova, L. Gorton and J. J. Calvente, J. Phys. Chem. B, 2007, 111, 469–477 CrossRef CAS PubMed.
- T. Vidakovic-Koch, V. K. Mittal, T. Q. N. Do, M. Varnicic and K. Sundmacher, Electrochim. Acta, 2013, 110, 94–104 CrossRef CAS PubMed.
- I. Ivanov, T. Vidakovic-Koch and K. Sundmacher, J. Power Sources, 2011, 196, 9260–9269 CrossRef CAS PubMed.
- I. Ivanov, T. Vidakovic-Koch and K. Sundmacher, J. Electroanal. Chem., 2013, 690, 68–73 CrossRef CAS PubMed.
- W. Jia, C. Jin, W. Xia, M. Muhler, W. Schuhmann and L. Stoica, Chem.–Eur. J., 2012, 18, 2783–2786 CrossRef CAS PubMed.
- C. Gomez, S. Shipovskov and E. E. Ferapontova, J. Renewable Sustainable Energy, 2010, 2, 013103-1–013103-12 Search PubMed.
- W. Z. Jia, S. Schwamborn, C. Jin, W. Xia, M. Muhler, W. Schuhmann and L. Stoica, Phys. Chem. Chem. Phys., 2010, 12, 10088–10092 RSC.
- T. Ruzgas, E. Csoregi, J. Emneus, L. Gorton and G. MarkoVarga, Anal. Chim. Acta, 1996, 330, 123–138 CrossRef CAS.
- T. Q. N. Do, M. Varnicic, R. Hanke-Rauschenbach, T. Vidakovic-Koch and K. Sundmacher, Electrochim. Acta, 2014, 137, 616–626 CrossRef CAS PubMed.
- B. Limoges, J. M. Saveant and D. Yazidi, J. Am. Chem. Soc., 2003, 125, 9192–9203 CrossRef CAS PubMed.
- J. Filip, J. Sefcovicova, P. Gemeiner and J. Tkac, Electrochim. Acta, 2013, 87, 366–374 CrossRef CAS PubMed.
- T. Tamaki and T. Yamaguchi, Ind. Eng. Chem. Res., 2006, 45, 3050–3058 CrossRef CAS.
- P. Gode, F. Jaouen, G. Lindbergh, A. Lundblad and G. Sundholm, Electrochim. Acta, 2003, 48, 4175–4187 CrossRef CAS.
- H. P. Erickson, Biol. Proced. Online, 2009, 11, 32–51 CrossRef CAS PubMed.
- M. ElKaoutit, A. H. Naggar, I. Naranjo-Rodriguez, M. Dominguez and J. de Cisneros, Synth. Met., 2009, 159, 541–545 CrossRef CAS PubMed.
- Y. H. Song, L. Wang, C. B. Ren, G. Y. Zhu and Z. Li, Sens. Actuators, B, 2006, 114, 1001–1006 CrossRef CAS PubMed.
- J. L. Zhang, F. Zhang, H. J. Yang, X. L. Huang, H. Liu, J. Y. Zhang and S. W. Guo, Langmuir, 2010, 26, 6083–6085 CrossRef CAS PubMed.
- K. Besteman, J. O. Lee, F. G. M. Wiertz, H. A. Heering and C. Dekker, Nano Lett., 2003, 3, 727–730 CrossRef CAS.
- K. De Wael, S. Van Vlierberghe, H. Buschop, P. Dubruel, B. Vekemans, E. Schacht, L. Vincze and A. Adriaens, Surf. Interface Anal., 2009, 41, 389–393 CrossRef CAS.
- J. D. Zhang, Q. J. Chi, S. J. Dong and E. K. Wang, Bioelectrochem. Bioenerg., 1996, 39, 267–274 CrossRef CAS.
- J. H. Wang, L. W. Ruddock and A. E. G. Cass, Biosens. Bioelectron., 1994, 9, 647–655 CrossRef CAS.
- H. P. Lu, L. Y. Xun and X. S. Xie, Science, 1998, 282, 1877–1882 CrossRef CAS.
- M. Xiong, B. Gu, J.-D. Zhang, J.-J. Xu, H.-Y. Chen and H. Zhong, Biosens. Bioelectron., 2013, 50, 229–234 CrossRef CAS PubMed.
- D. Olea, P. Moreau and C. Faure, J. Electroanal. Chem., 2007, 605, 125–135 CrossRef CAS PubMed.
- A. Uygun, L. Oksuz, S. Chowdhury and V. Bhethanabotla, Mater. Sci. Eng., C, 2010, 30, 868–872 CrossRef CAS PubMed.
- A. P. Hsiao and M. J. Heller, J. Biomed. Biotechnol., 2012, 2012, 178487 Search PubMed.
- S. E. Rosenwald, W. B. Nowall, N. Dontha and W. G. Kuhr, Anal. Chem., 2000, 72, 4914–4920 CrossRef CAS.
- B. Wetzl, M. Gruber, B. Oswald, A. Durkop, B. Weidgans, M. Probst and O. S. Wolfbeis, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2003, 793, 83–92 CrossRef CAS.
- T. Ruzgas, L. Gorton, J. Emneus and G. Markovarga, J. Electroanal. Chem., 1995, 391, 41–49 CrossRef.
- R. A. Sheldon, Org. Process Res. Dev., 2011, 15, 213–223 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra07495e |
| This journal is © The Royal Society of Chemistry 2014 |