
Pyrene-based D–π–A dyes that exhibit solvatochromism and high fluorescence brightness in apolar solvents and water†
Abstract
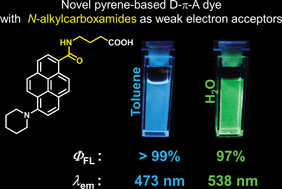
A novel pyrene-based D–π–A dye consisting of piperidine (D) and a secondary N-alkyl carboxamide (A) was prepared. This dye showed solvatochromism and bright fluorescence in solvents with a wide range of polarities, including water. Such emission properties are derived from the role of the secondary N-alkyl carboxamide group as both a weak acceptor and a stabilizer of the n electrons on the carbonyl group.
 Please wait while we load your content...
Please wait while we load your content...

