
Experimental and DFT study on the indium-mediated synthesis of benzophenones via arylstannanes†
Marcos J. Lo Fiego,
Viviana B. Dorn‡
*,
Mercedes A. Badajoz,
María T. Lockhart* and
Alicia B. Chopa§
INQUISUR, Departamento de Química, Universidad Nacional del Sur, Avenida Alem 1253, 8000 Bahía Blanca, Argentina. E-mail: lockhart@criba.edu.ar; vdorn@uns.edu.ar; Fax: +54 291 459 5187; Tel: +54 291 459 5187
First published on 18th September 2014
Abstract
Experimental results of the solvent-free, indium-promoted reaction of aroyl chlorides with arylstannanes showed a narrow scope; its efficiency depends both on the extent of methylation in the latter and on the nature, number and position of the substituents in the former. With the purpose of explaining experimental results, a theoretical analysis with DFT methods was performed for a set of selected cases.
Introduction
Ketones are vital building blocks in organic synthesis as well as an important functionality found in pharmaceutical compounds. The Friedel–Crafts (F–C) acylation of aromatic compounds is the most frequent route for the synthesis of aromatic ketones.1 However, it has intrinsic limitations such as the substituent-directing effects, the reactivity substrate requirements and the fact that recovery and recycling of the catalyst is seldom possible after aqueous work-up and a large amount of toxic waste is generated. On the other hand, Pd-catalyzed cross-coupling reactions of acyl halides with organometallic reagents provide a direct procedure for the synthesis of isomeric ketones;2 a drawback is that Pd-catalysts are expensive and they are not recovered. Arylstannanes are valuable reagents for the regiospecific generation of C–C bonds. In the last years, we have been involved in the synthesis of arylstannanes3 as well as in their application as intermediates in organic synthesis4 focused, especially, in the development of new procedures for the regiospecific synthesis of aryl ketones through catalyst-free processes.4b–eThe advance of indium-mediated synthetic methods has grown up in the recent literature due to the special properties of indium metal.5 It is unaffected by air, moisture or oxygen at ambient temperature and, most importantly, the element itself is without any apparent toxicity. In this respect, we have established that indium metal is a promoter of the solvent-free reaction of bulky aroyl chlorides with bulky arylstannanes, in the synthesis of severely hindered benzophenones;4e moreover, we have also established that it is a promoter of the solvent-free reaction of alkanoyl chlorides with arylstannanes, in the synthesis of primary, secondary and tertiary alkyl aryl ketones.4h The reactions are carried out under mild and neutral conditions, being their striking advantages (i) the use of no solvent, (ii) readily available, non-toxic and reusable metal catalyst; (iii) accessible substrates and reactants and (iv) the specificity of the reaction. On the latter point, it is worth noting that, whether the reaction proceeds via a free-radical or ionic mechanism, the arylstannane plays a key role in regards to the regioselectivity of the benzophenones synthesis. The high ipso reactivity makes it possible to carry out aromatic substitution under mild conditions overriding the normal directive effect of substituents. This is ascribed mainly to the stabilization of the cyclohexadienyl radical or cation intermediates by hyperconjugation between the singly occupied or unoccupied 2p orbital and the β-C–Sn bond (often referred as β-effect). Furthermore, unlike the corresponding homolytic substitution of hydrogen, the stannyl radical should be rapidly lost from the cyclohexadienyl intermediate without the need for any oxidant.6 In this connection we sought to establish the scope of this solvent free approach, with the ultimate goal of developing a general method for the synthesis of benzophenones. Thus, we initiated a systematic study of the reaction between a series of representative benzoyl chlorides (Fig. 1, 1a–g) with a series of arylstannanes (Fig. 1, 2a–d) and herein, we report the unexpected experimental results and a possible theoretical interpretation of this reaction.
 | ||
| Fig. 1 Starting materials: benzoyl chlorides and arylstannanes. | ||
Results and discussion
We initiated our studies with the reaction between benzoyl chloride (1a) and tributylphenylstannane (2a) as a model system. Unexpectedly, after long reaction times (24 h), even traces of the desired ketone were not detected although working at higher temperatures (50–100 °C) or employing an excess of 1a.Next, and based on our previous experience where arylstannanes containing electron-releasing substituents were proved to be adequate substrates under this protocol,4e,h we carried out a series of reactions between 1a and arylstannanes 2b–d (Table 1, entries 1 to 3). The results obtained showed that working at 80 °C (24 h),7 meanwhile 2b was unreactive, the reaction of 2c and 2d with 1a yielded the corresponding ketones 3ac and 3ad, although in really low yields (traces and 20%, respectively), and substantial amounts of reactant were recovered. It should be mentioned that 3ad was obtained together with the undesired isomeric ketone (2,4-dimethylphenyl)phenyl methanone (5%), product of the direct aroylation of the aromatic ring followed by protodestannylation. Likewise, the reaction of arylstannanes 2a to 2d with (4-methoxy)benzoyl chloride (1b) gave the corresponding ketones, (traces up to 25%) (Table 1, entries 4 to 7). In contrast, the reaction of 2d with (4-nitro)benzoyl chloride (1c) was negative even after 72 h (Table 1, entry 8).
|
|||||
|---|---|---|---|---|---|
| Entrya | ArCOCl | Ar′SnR3 | Time (h) | ArCOAr′ | Yieldb (%) |
| a All reactions were conducted at 80 °C (oil bath) in solventless conditions using 1.2 equiv. of 1, 1.0 equiv. of 2 and 1.0 equiv. of indium metal.b Determined by GC-MS (using tetradecane as internal standard).c Together with the regioisomer [(2,4-dimethylphenyl)phenyl methanone, 5%].d Traces of mesitylene were detected (GC-MS) and large amounts of unreacted starting materials were recovered.e Together with the regioisomer [(3,4-dimethoxyphenyl)(2,4-dimethylphenyl)methanone, 4%].f Together with the regioisomer [(3,4,5-trimethoxyphenyl)(2,4-dimethylphenyl)methanone, 10%].g Ref. 4e.h Performed with 0.2 equiv. of indium metal. | |||||
| 1 | 1a | 2b | 24 |  |
0 |
| 2 | 1a | 2c | 24 | 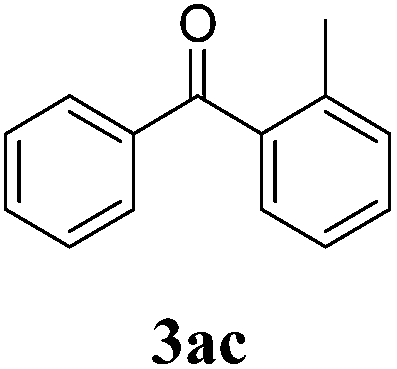 |
Traces |
| 3 | 1a | 2d | 24 |  |
20c |
| 4 | 1b | 2a | 24 |  |
Traces |
| 5 | 1b | 2b | 24 |  |
4 |
| 6 | 1b | 2c | 24 |  |
9 |
| 7 | 1b | 2d | 24 |  |
25 |
| 8 | 1c | 2d | 72 |  |
0 |
| 9 | 1d | 2d | 2 |  |
6d |
| 10 | 1e | 2d | 2 |  |
48d,e |
| 11 | 1f | 2d | 2 |  |
32d,f |
| 12g | 1g | 2d | 2 | 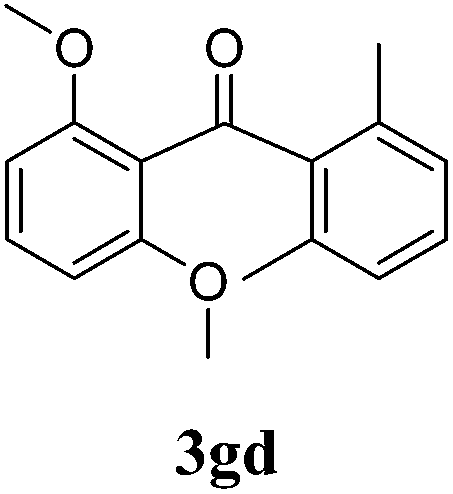 |
55 |
| 13g,h | 1g | 2d | 2 | 3gd | 49 |

These preliminary results showed that the nature of the benzoyl chloride is an important parameter in these reactions and that a π-electron releasing group attached to the aromatic ring increased its reactivity (entries 1–3 vs. 5–7 and entry 7 vs. 8). Furthermore, the reactions carried out with arylstannane 2d gave better yields (entries 3 and 7).
Based on these outcomes we considered it interesting to study the reactivity of arylstannane 2d towards different commercially available benzoyl chlorides containing two or three methoxy groups in diverse positions, such as (3,5-dimethoxy)benzoyl chloride (1d), (3,4-dimethoxy)benzoyl chloride (1e) and (3,4,5-trimethoxy)benzoyl chloride (1f). The reactions, which were quenched at 2 h in order to compare results with those we have formerly reported,4e gave the corresponding ketones 3dd, 3ed and 3fd with 6%, 48% and 32% yield, respectively (Table 1, entries 9 to 11). In a previous work we have informed the synthesis of the bulky ketone 3gd (55%; Table 1, entry 12) under a similar protocol.4e So, it is evident that the effectiveness of the reaction depends mainly on the relative position of the methoxy groups in the aromatic ring rather than their number. Thus, isomers 1e (3,4-dimethoxy) and 1g (2,6-dimethoxy) are more reactive than isomer 1d (3,5-dimethoxy) and even than 1f (3,4,5-trimethoxy).
It is important to note that a control experiment showed that no reaction occurred in the absence of In(0). Besides, it is known that, because of its low first ionization potential, In(0) is capable of promoting single-electron transfer (SET) processes.8 To prove whether acyldestannylations proceeded through a polar or a free radical pathway, we performed the reaction of 1f and 2d in the presence of 0.5 equiv. of di-tert-butyl nitroxide (DTBN) as radical scavenger. Compared with a blank reaction, we noticed that the addition of DTBN had a dramatic retardation effect and only traces of ketone 3fd were detected recovering the starting substrate. On the basis of these results, we believe that the reaction proceeds initially through a SET from In(0) to the aroyl chloride with generation of an acyl radical and In(I) chloride salt. The acyl radical thus formed reacts with the arylstannane through a homolytic ipso aromatic substitution affording the ketone (Scheme 1).4e The high ortho-selectivity make it reasonable to speculate that the formation of the cyclohexadienyl radical intermediate is the rate determining step (RDS) which is conditioned by the degree of fragmentation of the acyl radical anion and the reactivity of the corresponding acyl radical (Scheme 1).
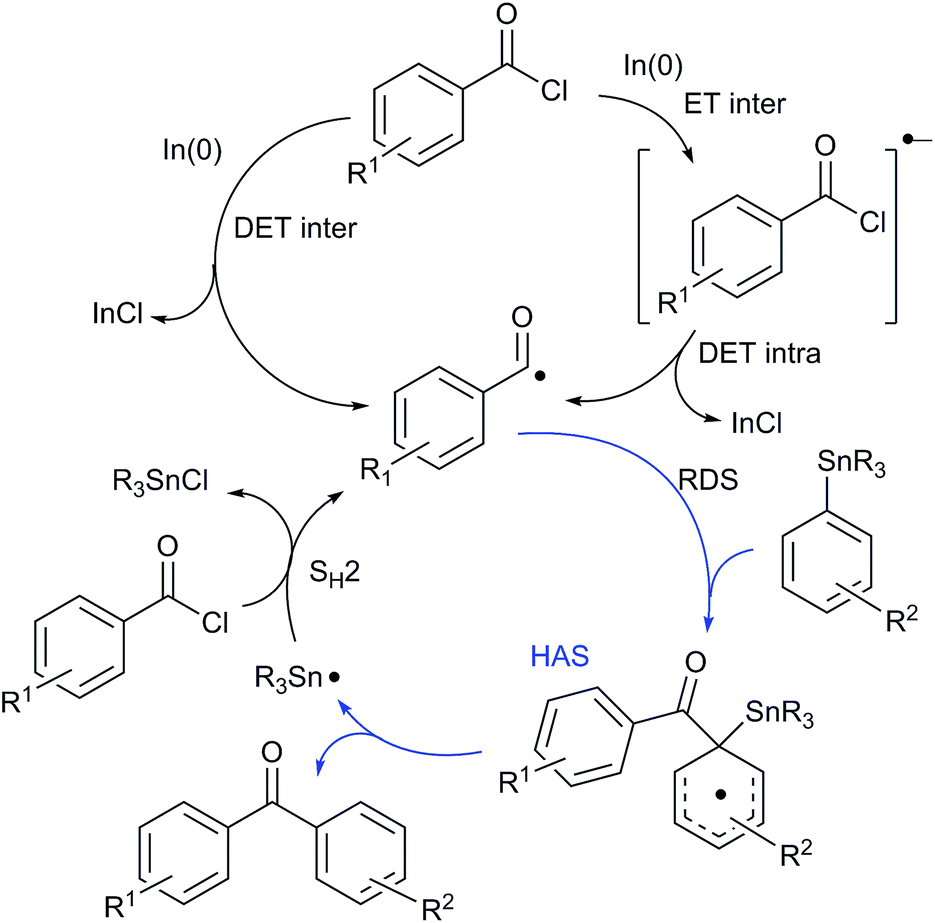 | ||
| Scheme 1 | ||
The unexpected results together with the fact that the reactions proceed through an electron transfer (ET) pathway generating an acyl radical (Scheme 1), prompted us to study, in more detail, the ET process involved using density functional theory (DFT) calculations. For this purpose, we simplified the reactive system and theoretically studied the neutral acyl chlorides and the corresponding radical anions of compounds 1a, 1c and the dimethoxybenzoyl chlorides isomers 1d and 1g as representative compounds. The density functional theory9 calculations were performed with the B3LYP10 functional and the 6-31+G* basis set, which is known to be an appropriate methodology for the theoretical studies of electron affinities11 and electronic properties of radicals12 and radical anions.13
Adiabatic electron affinity (AEA) can be used as a magnitude of the reactivity of a molecule toward a SET process. Table 2 shows AEA of the neutral benzoyl chlorides, obtained as the difference in the energies between the neutral molecule and the radical anion with full geometry optimization of both species, including the zero point energy (ZPE) corrections (eqn (1)).11b–d As expected, benzoyl chlorides showed differences in energy.
| AEA = E(optimized neutral) − E(optimized radical anion) | (1) |
| Benzoyl chlorides | Adiabatic EAs energy (eV) |
|---|---|
 |
2.37 |
 |
0.98 |
 |
0.89 |
| 1g | 0.58 |
The presence of an electron-withdrawing group attached to the aromatic ring (1c) increases the stability of the π-system.14 Evidence of this is the shortening of the C–NO2 bond in the radical anion (1.40 Å) with respect to the neutral (1.48 Å).13a,14b
Thus, the electron transfer (ET-inter) process is favored, by increasing the adiabatic electron affinity of the substrate. The unreactivity of derivative 1c is related to the distribution of spin density,13a,d,14b a relevant factor for reactions that proceed through ET processes.15 Unlike radical anions 1d and 1g, once the corresponding radical anion is generated in 1c, the unpaired electron is located mainly on the nitro function rather than on the carbon center of the acyl group (Fig. 2); therefore, even when the 1c radical anion was efficiently generated, the expected fragmentation does not take place and the acyl radical is not generated.
 | ||
| Fig. 2 Gas phase B3LYP/6-31+G* spin density (orange, isosurface of 0.02) for 1c˙−, 1d˙− and 1g˙−. | ||
The situation for isomers 1d and 1g is different; although both have, as expected, similar electron affinities, their behavior towards the most reactive arylstannane 2d is completely different. Thus, while 1g gives the ketone in a good yield (55%), 1d gives the product in only 6% yield. Taking this in account, the goal of this computational study is to analyze the corresponding radical anions, as intermediates in the substitution, bearing in mind the spin density and fragmentation process as possible factors responsible for the observed differences. As can be seen from Fig. 2, there are no differences in the unpaired electron of both radical anions. With respect to the fragmentation process, once the neutral acyl chloride gain an electron from In(0) (inter-ET), the reaction may follow two mechanisms:13a,14b (i) a concerted dissociative pathway where the CO–Cl bond breaks as the benzoyl radical anion is being formed (inter-DET); (ii) a stepwise mechanism, where takes place an intramolecular dissociative ET (intra-DET) from the π system to the σ* CO–Cl bond once the benzoyl radical anion is formed (Scheme 1). Considering that, we evaluated the potential energy surfaces (PES) for the dissociation of both intermediates. Our results indicate that the formation of the benzoyl radical from 1d occurs exothermically (stepwise mechanism) through a radical anion intermediate, being the activation energy 8.1 kcal mol−1 (ref. 17) (Fig. 3). On the other hand, the fragmentation of the radical anion 1g takes place without activation energy generating the corresponding radical by a spontaneous dissociative electron transfer (inter-DET) (Scheme 1).
 | ||
| Fig. 3 Gas phase B3LYP/6-31+G* potential energy profile for dissociation of 1d˙−.20 Spin densities are shown in orange (isosurface of 0.02). | ||
As can be seen from Fig. 4, the main geometric difference between both radical anions isomers is that radical anion 1g shows an out of plane distortion from the optimal sp2 dihedral angle Cl–CO–C1–C2 from 0° to −20.5°,18 probably due to steric repulsion with the ortho-methoxy substitutents. The SOMO MO shows that this distortion could facilitate the cleavage of this radical anion,14b,19 being more efficient the intra-DET process since this spatial geometric arrangement increases the interaction and the overlap of the π and σ* orbitals in comparison to the planar radical anion 1d.
 | ||
| Fig. 4 B3LYP/6-31+G* geometry of 1g˙− and SOMO MOs (orange and blue, isosurface of 0.02) of 1g˙−and 1d˙−. | ||
Experimental section
General experimental methods
All reactions were carried out under a dry nitrogen atmosphere. Acid chlorides were commercially available and fractionally distilled under nitrogen or recrystallized from hexane before use. Aryltributylstannanes 2a–c were prepared by transmetallation of the appropriate Grignard reagents with tributyltin chloride in anhydrous THF. Aryltrimethylstannane 2d was obtained from the corresponding commercial aryl chloride by photostimulated reaction with Me3SnNa in liquid ammonia, according to the literature procedures.21The products were identified unambiguously by comparing their retention times and mass spectra with those of authentic compounds obtained by known procedures,4e using a GC/MS instrument (HP5-MS capillary column, 30 m × 0.25 mm × 0.25 μm) equipped with 5972 mass selective detector operating at 70 eV (EI). Program: 50 °C for 2 min with increase 10 °C min−1 to 280 °C.
General procedure for indium-mediated reactions
In a flame dried Schlenk tube (fitted with a teflon plug valve) 1.2 mmol of acid chloride 1 was added to a stirred mixture of 1.0 mmol of arylstannane 2 and indium powder (1.0 mmol) under a nitrogen gas stream. The system was purged with nitrogen by means of three pump-fill cycles and then the heterogeneous reaction mixture was stirred at 80 °C (oil bath) for the time indicated in Table 1. After addition of 10% (m/v) solution of NaOH (2 mL) and 10 μL of tetradecane, the mixture was stirred at room temperature for 15 min and then diluted with DCM (5 mL). The organic phase was successively washed with water and brine, dried over Na2SO4, filtered and analyzed by GC and GC/MS.Computational procedure
The calculations were performed with Gaussian09.25 The initial conformational analysis of selected compounds was performed with the semiempirical AM1 method. The geometry of the most stable conformers thus obtained was used as starting point for the B3LYP studies of the corresponding benzoyl chlorides and their radical anions. Zero point energy and thermal energy were computed at the 6-31+G* level and scaled by a factor 0.986 (ref. 26) for adiabatic EAs and thermodynamic quantities. The potential energy surface was inspected through a scan of the distinguished reaction coordinate. That is, by elongating the C(O)–Cl bond (steps of 0.01 Å each) at B3LYP/6-31+G* level with full optimization for the remainder degrees of freedom. The energy profile obtained is shown in Fig. 3. The characterization of all stationary points was done by Hessian matrix calculations of geometries obtained with full optimization for a minimum and by using the QST2 methodology for a transition state. All calculations were performed in gas phase.27Conclusions
Despite the limited scope of this methodology, our results are a contribution to the research related to the action of metallic indium as a promoter of free-radical reactions in organic synthesis. We have observed that the effectiveness of the synthesis of benzophenones, through the indium-promoted solvent-free reaction of aroyl chlorides with arylstannanes, depends not only on the extent of methylation in the latter but, in a very peculiar way, on the nature, number and position of the substituents in the former. A good agreement between DFT calculation and the experimental results was found for the analyzed cases. In this way the B3LYP functional and the 6-31+G* basis set demonstrated to be an adequate and computationally economic methodology for studying reactive systems that imply radicals and radical anions involved in ET processes.Acknowledgements
We are grateful for financial support provided by CONICET (Consejo Nacional de Investigaciones Científicas y Técnicas), ANPCyT (Agencia Nacional de Promoción Científica y Tecnológica), CIC (Comisión de Investigaciones Científicas) and SGCyT-UNS (Secretaría General de Ciencia y Técnica – Universidad Nacional del Sur) from Argentina. CONICET is thanked for a research fellowship to MLF. The authors would like to thanks Prof. A. B. Pierini for her feedback and advisory with this manuscript.Notes and references
- (a) M. B. Smith and J. March, March's Advanced Organic Chemistry, John Wiley, New York, 6th edn, 2007 Search PubMed; (b) R. C. Larock, Comprehensive Organic Transformations, Wiley, VCH, New York, 1999 Search PubMed.
- For a review see: K. Dieter, Tetrahedron, 1999, 55, 4177 CrossRef.
- (a) A. B. Chopa, M. T. Lockhart and G. F. Silbestri, Organometallics, 2000, 19, 2249 CrossRef CAS; (b) A. B. Chopa, M. T. Lockhart and G. F. Silbestri, Organometallics, 2001, 20, 3358 CrossRef CAS; (c) A. B. Chopa, M. T. Lockhart and V. B. Dorn, Organometallics, 2002, 21, 1425 CrossRef CAS; (d) A. B. Chopa, M. T. Lockhart and G. F. Silbestri, Organometallics, 2002, 21, 5874 CrossRef CAS; (e) G. F. Silbestri, M. J. Lo Fiego, M. T. Lockhart and A. B. Chopa, J. Organomet. Chem., 2010, 695, 2578 CrossRef CAS; (f) G. F. Silbestri, M. T. Lockhart and A. B. Chopa, ARKIVOC, 2011, 210 CAS; (g) V. B. Dorn, G. F. Silbestri, M. T. Lockhart, A. B. Chopa and A. B. Pierini, New J. Chem., 2013, 37, 1150 RSC.
- (a) A. B. Chopa, G. F. Silbestri and M. T. Lockhart, J. Organomet. Chem., 2005, 690, 3865 CrossRef CAS; (b) G. F. Silbestri, R. Bogel Masson, M. T. Lockhart and A. B. Chopa, J. Organomet. Chem., 2006, 691, 1520 CrossRef CAS; (c) M. J. Lo Fiego, M. A. Badajoz, G. F. Silbestri, M. T. Lockhart and A. B. Chopa, J. Org. Chem., 2008, 73, 9184 CrossRef CAS PubMed; (d) M. J. Lo Fiego, M. T. Lockhart and A. B. Chopa, J. Organomet. Chem., 2009, 694, 3674 CrossRef CAS; (e) M. J. Lo Fiego, G. F. Silbestri, A. B. Chopa and M. T. Lockhart, J. Org. Chem., 2011, 76, 1707 CrossRef CAS PubMed; (f) C. E. Domini, G. F. Silbestri, B. Fernández Band and A. B. Chopa, Ultrason. Sonochem., 2012, 19, 410 CrossRef CAS PubMed; (g) M. Luong, C. E. Domini, G. F. Silbestri and A. B. Chopa, J. Organomet. Chem., 2013, 723, 43 CrossRef CAS; (h) M. J. Lo Fiego, M. A. Badajoz, C. E. Domini, A. B. Chopa and M. T. Lockhart, Ultrason. Sonochem., 2013, 20, 826 CrossRef CAS PubMed.
- For reviews see: (a) B. C. Ranu, Eur. J. Org. Chem., 2000, 2347 CrossRef CAS; (b) V. Nair, S. Ros, C. N. Jayan and B. S. Pillia, Tetrahedron, 2004, 60, 1959 CrossRef CAS; (c) J. Augé, N. Lubin-Germain and J. Uziel, Synthesis, 2007, 1739 CrossRef; (d) J. S. Yadav, A. Antony, J. George and B. V. Reddy, Eur. J. Org. Chem., 2010, 591 CrossRef CAS; (e) A. K. Singh, Synlett, 2013, 24, 1457 CrossRef.
- See “Metals as surrogates for hydrogen in organic chemistry: anything hydrogen can do, a metal can do better”, A. G. Davies, J. Chem. Soc., Perkin Trans. 1, 2000, 1997 RSC.
- This temperature is required to keep an efficient stirring of the reaction mixture.
- G. Marin, A. L. Braga, A. S. Rosa, F. Z. Galetto, R. A. Burrow, H. Gallardo and M. W. Paixao, Tetrahedron, 2009, 65, 4614 CrossRef CAS.
- W. Kohn and I. J. Sham, Phys. Rev., 1965, 140, A1133 CrossRef.
- (a) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS; (b) A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS PubMed; (c) E. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200 CrossRef.
- (a) S. E. Boesch, A. K. Grafton and R. A. Wheeler, J. Phys. Chem., 1996, 100, 10083 CrossRef CAS; (b) D. M. A. Vera and A. B. Pierini, Phys. Chem. Chem. Phys., 2004, 6, 2899 RSC; (c) M. Puiatti, D. M. A. Vera and A. B. Pierini, Phys. Chem. Chem. Phys., 2008, 10, 1394 RSC; (d) M. Puiatti, D. M. A. Vera and A. B. Pierini, Phys. Chem. Chem. Phys., 2009, 11, 9013 RSC and references therein.
- L. E. Peisino and A. B. Pierini, J. Org. Chem., 2013, 78, 4719 CrossRef CAS PubMed.
- (a) A. B. Pierini and D. M. A. Vera, J. Org. Chem., 2003, 68, 9191 CrossRef CAS PubMed; (b) D. M. A. Vera and A. B. Pierini, Phys. Chem. Chem. Phys., 2004, 6, 2899 RSC; (c) J. G. Uranga, D. M. A. Vera, A. N. Santiago and A. B. Pierini, J. Org. Chem., 2006, 71, 6596 CrossRef CAS PubMed; (d) X. Asensio, A. González-Lafont, J. Marquet, J. M. Lluch and M. Geoffroy, J. Mol. Struct.: THEOCHEM, 2009, 913, 228 CrossRef CAS.
- (a) E. R. Vorpagel, A. Streitwieser Jr and S. D. Alexandratos, J. Am. Chem. Soc., 1981, 103, 3777 CrossRef CAS; (b) A. B. Pierini, J. S. Duca and M. A. Vera, J. Chem. Soc., Perkin Trans. 2, 1999, 1003 RSC.
- (a) R. A. Rossi and R. H. de Rossi, Aromatic Substitution by the SRN1 Mechanism, American Chemical Society Monograph Series, 1983, p. 178 Search PubMed; (b) V. B. Dorn, M. A. Badajoz, M. T. Lockhart, A. B. Chopa and A. B. Pierini, J. Organomet. Chem., 2008, 693, 2458 CrossRef CAS; (c) F. Nador, Y. Moglie, A. Ciolino, A. Pierini, V. Dorn, M. Yus, F. Alonso and G. Radivoy, Tetrahedron Lett., 2012, 53, 3156 CrossRef CAS.
- Better calculations level has been attempted, enlarging the basis set to 6-31+G** and 6-31++G**. However, not substantial changes have been observed. The adiabatic electron affinity for 1c was 2.37 eV, 2.38 eV and 2.38 eV using 6-31+G*, 6-31+G** and 6-31++G** respectively.
- Similar results were obtained with a better calculation level for the AE, 8.1 and 8.0 kcal mol−1 after applying 6-31+G** and 6-31++G**, respectively.
- 20.7° and 20.8° were the values obtained for the dihedral angle Cl–CO–C1–C2 with 6-31+G** and 6-31++G**, respectively.
- A. B. Pierini and J. S. Duca, J. Chem. Soc., Perkin Trans. 2, 1995, 1821 RSC.
- It should be mentioned that, when the 6-31+G** and 6-31++G** basis set was applied in the calculations, the spin density remained almost unchanged.
- C. C. Yammal, J. C. Podestá and R. A. Rossi, J. Org. Chem., 1992, 57, 5720 CrossRef CAS.
- J. R. Schmink and S. W. Krska, J. Am. Chem. Soc., 2011, 133, 19574 CrossRef CAS PubMed.
- B. M. O'Keefe, N. Simmons and S. F. Martin, Org. Lett., 2008, 10, 5301 CrossRef PubMed.
- W. J. Wu, T. C. Lin, T. Takahashi, F. Y. Tsai and C. Y. Mou, ChemCatChem, 2013, 5, 1011 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- I. M. Alecu, J. Zheng, Y. Zhao and D. G. Truhlar, J. Chem. Theory Comput., 2010, 6, 2872 CrossRef CAS.
- Although we do not know the effective polarity of the reaction medium, we evaluate the dependence of the chemical reactivity associated with derivatives 1d and 1g in condensed phase using the PCM model (S. Miertus, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117. S. Miertus and J. Tomasi, Chem. Phys., 1982, 65, 239. M. Cossi, V. Barone, R. Cammi and J. Tomasi, Chem. Phys. Lett., 1996, 255, 327.) implemented in Gaussian09 in three different solvents (acetone (ε = 20.7), o-dichlorobenzene (ε = 9.9), chloroform (ε = 4.7)) thus covering a wide range of dielectric constants. The evaluation of the obtained adiabatic electron affinities and activation energy (reported in the ESI†) allow us to conclude that the gas phase calculations are a good approach in order to explain the reactivity of this experimental system.
Footnotes |
| † Electronic supplementary information (ESI) available: xyz coordinates and total energies in atomic units for all of the calculated structures and copy of 1H and 13C NMR spectra of unknown compounds 3ed, 3dd and 3fd. See DOI: 10.1039/c4ra07305c |
| ‡ Member of CONICET. |
| § Member of CIC. |
| This journal is © The Royal Society of Chemistry 2014 |