
Non-aqueous sol–gel synthesis through a low-temperature solvothermal process of anatase showing visible-light photocatalytic activity†
Abstract
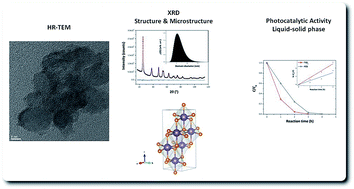
A novel, facile method based on a non-aqueous sol–gel solvothermal process has been developed to synthesise spherical TiO2 nanoparticles (NPs) in one pot. The reaction between titanium(IV) tert-butoxide (Ti[OC(CH3)3]4) and benzyl alcohol was a simple process, which resulted in the formation of highly crystalline titania NPs with a small size of only 6 nm, and with a correspondingly high surface area. The chemical formation mechanism of the metal oxide NPs has been proposed, and the degree of surface hydroxyls (–OH groups) has been examined. The products of the synthesis were characterised by X-ray powder diffraction (XRPD) using the advanced whole powder pattern modelling (WPPM) method, high-resolution transmission electron microscopy (HR-TEM), thermo-gravimetric analysis (TGA), UV-visible diffuse reflectance spectroscopy (DRS), Fourier transform infrared spectroscopy (FT-IR), and nuclear magnetic resonance (NMR) spectroscopy. The photocatalytic activity (PCA) was evaluated in both the liquid–solid phase, by monitoring the degradation of an organic dye (methylene blue (MB)) under UV-light irradiation, and in the gas–solid phase, by following the degradation of 2-propanol under UV- and visible-light exposures. The synthesized titania powders not only exhibited excellent photocatalysis in the liquid–solid phase (under UV irradiation), but also possessed a superior PCA in the gas–solid phase under a visible-light exposure. The effects on the PCA of the very small crystalline domain size, surface composition and the presence of organic molecules due to the synthesis process of the TiO2 NPs were shown to account for this behaviour.
 Please wait while we load your content...
Please wait while we load your content...

