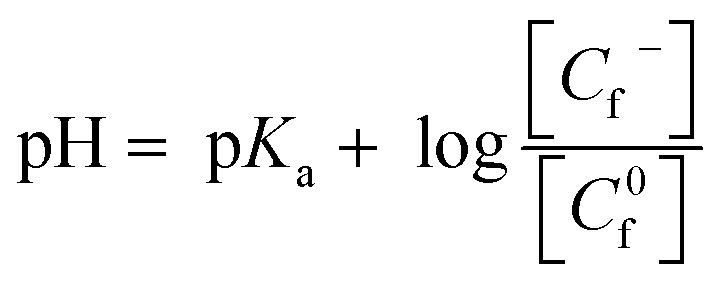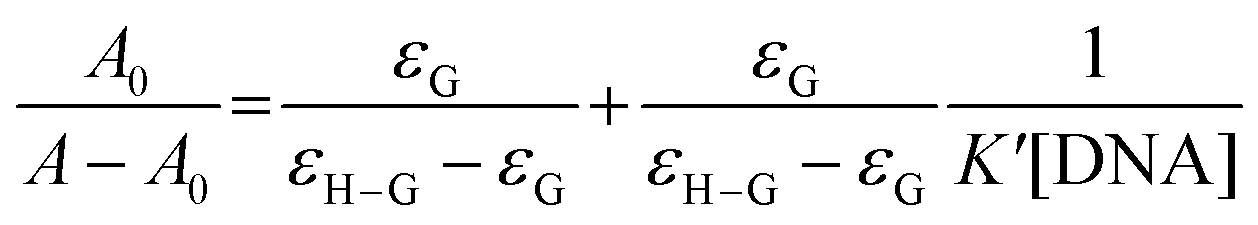DOI:
10.1039/C4RA07127A
(Paper)
RSC Adv., 2014,
4, 59344-59357
Synthesis, crystal structure, DNA interaction and in vitro anticancer activity of a Cu(II) complex of purpurin: dual poison for human DNA topoisomerase I and II†
Received
15th July 2014
, Accepted 24th October 2014
First published on 27th October 2014
Abstract
Although generation of reactive oxygen species (ROS) by anthracycline anticancer drugs is essential for anti-tumor activity, they make these drugs cardiotoxic. Metal–anthracyclines that generate relatively fewer ROS are however, effective antitumor agents. Purpurin (LH3), a hydroxy-9,10-anthraquinone, closely resembles doxorubicin, an established anthracycline drug. This molecule was chosen to study the extent to which simpler analogues are effective. A Cu(II) complex of LH3 [Cu(II)–(LH2)2] was synthesized to mimic the metal–anthracycline complexes. The crystal structure of [Cu(II)–(LH2)2] was determined by Rietveld refinement of PXRD data using an appropriate structural model developed on the basis of spectroscopic data. This is the first report on the crystal structure of any hydroxy-9,10-anthraquinone with a 3d-transition metal ion. The bond lengths and bond angles obtained by structural refinement corroborate those calculated by the DFT method. DNA binding of the complex was slightly better than purpurin. However, more importantly, unlike purpurin, binding constant values did not decrease with increasing pH of the medium. DNA relaxation assays show Cu(II)–(LH2)2 as a novel potent dual inhibitor of human DNA topoisomerase I and topoisomerase II enzymes. Cu(II)–(LH2)2 stabilizes covalent topoisomerase–DNA adducts both in vitro and within cancer cells. The cleavage assay keeps the complex well ahead of LH3 with regard to efficacy. These results paralleled those of cell growth inhibition and showed that the complex was more effective in killing ALL MOLT-4 cells than LH3, suggesting it targets topoisomerase enzymes within cells. The NADH dehydrogenase assay revealed further that the generation of superoxide was less in the case of the complex as compared to LH3.
1. Introduction
Anthracycline anticancer agents are currently in use in various forms of chemotherapy.1 The two most well known drugs in this series, adriamycin (doxorubicin, DOX) and daunomycin are extensively used in breast cancer and acute lymphoblastic leukemia (ALL). These compounds function as DNA topoisomerase inhibitors.1 DNA topoisomerases are essential nuclear enzymes found in cells that help to maintain DNA topology during processes like replication, transcription and recombination, ensuring the faithful segregation of chromosomes during cell division.1–3 The nuclei of mammalian cells contain two principal types of DNA topoisomerases, DNA topoisomerase I (topo I) and DNA topoisomerase II (topo II), which are molecular targets for several anticancer drugs.2,3 Topoisomerase inhibitors belong to two different classes. Class I inhibitors or topoisomerase poisons inhibit controlled strand rotation or the re-ligation step of the enzymatic reaction cycle forming enzyme linked DNA lesions that initiate cell cycle arrest, leading to apoptosis.4 Class II inhibitors or catalytic topoisomerase inhibitors inhibit DNA binding or the DNA nicking step of the enzyme-reaction cycle.5–8
The major concern on the use of anthracyclines is their associated cardiotoxicity and life-threatening heart damage.9–18 Studies on breast cancer itself reveal that ∼27% of patients suffer congestive heart failure later in life owing to anthracycline treatment.19,20 Reports have indicated that reactive oxygen species (ROS) produced by anthracyclines are mainly responsible for cardiotoxicity and that the hydroxy-9,10-anthraquinone moiety present in these molecules is the site of generation of ROS.1 At the same time, ROS is also essential for anticancer activity.21,22 Hence, there is a need to have a proper balance in the generation of ROS such that it maintains its anticancer activity but is not cardiotoxic. Lowering ROS generation to appropriate levels could be one approach to minimize the side effects of these drugs and their analogues.
Recent studies on hydroxy-9,10-anthraquinones and their metal complexes have shown remarkable similarities with anthracyclines, particularly with regard to physicochemical attributes, electrochemical behavior and biophysical interactions.23–25 The cardiotoxicity of anthracyclines was shown to decrease significantly when they form complexes with metal ions.26,27 Complexes of Cu(II) with anthracyclines generate relatively small amounts of ROS but are effective anti-cancer agents.26–29 With these facts in mind, our aim was to pick simple hydroxy-9,10-anthraquinones, prepare their complexes and compare them with similar complexes of anthracyclines.30 Purpurin (1,2,4-trihydroxy-9,10-anthraquinone), which closely resembles DOX, was chosen and a Cu(II) complex was prepared. Results obtained with this complex were compared with Cu(II) complexes of DOX with regard to semiquinone formation, DNA interaction and action on DNA topoisomerase enzymes to realize its potential as an anticancer agent.31
2. Experimental
2.1 Synthesis of a Cu(II) complex of LH3
Purpurin (LH3) of ∼96% purity was purchased from Sigma-Aldrich and further purified by re-crystallization from ethanol–water mixtures. The compound, being photosensitive, was stored in the dark. The complex was prepared by mixing Cu(NO3)2·3H2O and LH3 in the ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (metal to ligand stoichiometry was determined by the molar ratio method, ESI†). LH3 (0.09 mmol) and Cu(NO3)2·3H2O (0.045 mmol) were dissolved in a minimum volume of hot absolute ethanol and triple distilled water, respectively. The Cu(II) solution was added drop by drop to the yellowish-orange solution of LH3 taken in a beaker, with continuous stirring with the help of a temperature-controlled magnetic stirrer. The intense yellowish-orange color of LH3 changed almost immediately to dark brown upon addition of the aqueous Cu(II) solution. The solvent ratio was 2:1 ethanol–water. The reaction mixture was stirred for 2 hours at 40 °C. After 2 hours, it was kept overnight in the dark. A brown solid settled to the bottom of the beaker while the supernatant was yellow in color. The solution was filtered. The brown residue was washed with warm 10% ethanol solution. It was then purified from acetonitrile and dried in a vacuum desiccator. Utmost care was taken to prevent the formation of a polymeric 1:1 complex under alkaline conditions.32 The pH of the medium was maintained at ∼6.00 using sodium bicarbonate.
:2 (metal to ligand stoichiometry was determined by the molar ratio method, ESI†). LH3 (0.09 mmol) and Cu(NO3)2·3H2O (0.045 mmol) were dissolved in a minimum volume of hot absolute ethanol and triple distilled water, respectively. The Cu(II) solution was added drop by drop to the yellowish-orange solution of LH3 taken in a beaker, with continuous stirring with the help of a temperature-controlled magnetic stirrer. The intense yellowish-orange color of LH3 changed almost immediately to dark brown upon addition of the aqueous Cu(II) solution. The solvent ratio was 2:1 ethanol–water. The reaction mixture was stirred for 2 hours at 40 °C. After 2 hours, it was kept overnight in the dark. A brown solid settled to the bottom of the beaker while the supernatant was yellow in color. The solution was filtered. The brown residue was washed with warm 10% ethanol solution. It was then purified from acetonitrile and dried in a vacuum desiccator. Utmost care was taken to prevent the formation of a polymeric 1:1 complex under alkaline conditions.32 The pH of the medium was maintained at ∼6.00 using sodium bicarbonate.
Yield: 68%. Analytical calculation (%) for CuC28H18O12: C, 55.13; H, 2.95. Experimentally found: C, 54.81; H, 2.90. Cu(II) was estimated using the standard procedure.33 Analytical calculation (%) 10.42, experimentally found 10.34. UV-Vis spectra: λmax at 515 nm. MS (m/z): 630 [M + Na]+.
2.2 X-ray powder diffraction measurements and crystal structure of Cu(II)–(LH2)2
The X-ray powder diffraction data was collected on a Bruker D8 Advanced powder diffractometer using Cu-Kα radiation (λ = 1.5418 Å). The generator settings were 40 kV and 40 mA. The diffraction pattern was recorded at room temperature (21 °C) with step size 0.0199° (2θ) and a count time of 5 s per step over the 2θ range 5–80°. The X-ray powder diffraction pattern was indexed by NTREOR of EXPO 2009 package and the results indicate the sample crystallizes in a triclinic unit cell with a = 12.575 Å, b = 12.412 Å, c = 10.343 Å and α = 97.09°, β = 113.49°, γ = 62.38° [M(20) = 14, F(20) = 14(0.003767, 338)].34 The statistical analysis of powder patterns using the FINDSPACE of EXPO 2009 software package indicates that the most probable space group is P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The structure was solved by a global optimization method in direct-space using a Monte Carlo based simulated annealing technique (in parallel tempering mode), as implemented in the program FOX with the help of an initial structural model developed on the basis of spectroscopic information (provided in ESI†).35 The initial molecular geometry was optimized by MOPAC 2007 program and this optimized structural model was used as an input to the FOX program in order to obtain the atomic coordinates.35,36 The atomic coordinates thus obtained from FOX were used as the starting model for Rietveld refinement carried out by the GSAS software package with EXPGUI interface.37,38 The lattice parameters, background coefficients and profile parameters were refined. The background was described by the shifted Chebyshev function of the first kind with 24 points regularly distributed over the entire 2θ range. A fixed isotropic displacement parameter of 0.04 Å2 for all non-hydrogen atoms and 0.06 Å2 for hydrogen atoms was maintained. In the final stage of refinement, the preferred orientation correction was applied using the generalized spherical harmonic model of order 10.
. The structure was solved by a global optimization method in direct-space using a Monte Carlo based simulated annealing technique (in parallel tempering mode), as implemented in the program FOX with the help of an initial structural model developed on the basis of spectroscopic information (provided in ESI†).35 The initial molecular geometry was optimized by MOPAC 2007 program and this optimized structural model was used as an input to the FOX program in order to obtain the atomic coordinates.35,36 The atomic coordinates thus obtained from FOX were used as the starting model for Rietveld refinement carried out by the GSAS software package with EXPGUI interface.37,38 The lattice parameters, background coefficients and profile parameters were refined. The background was described by the shifted Chebyshev function of the first kind with 24 points regularly distributed over the entire 2θ range. A fixed isotropic displacement parameter of 0.04 Å2 for all non-hydrogen atoms and 0.06 Å2 for hydrogen atoms was maintained. In the final stage of refinement, the preferred orientation correction was applied using the generalized spherical harmonic model of order 10.
2.3 Computational details
Full geometry optimizations were carried out at the (U)B3LYP levels using the density functional theory method with Gaussian 09 (revision A.02).39–41 All elements were assigned by the LanL2DZ basis set with effective core potential.42,43 Stationary-point calculations were carried out using the output coordinates from the geometry optimization calculations. GaussSum was used to calculate the fractional contributions of various groups to each molecular orbital.44,45 No symmetry constraints were imposed during structural optimizations and the nature of the optimized structures and energy minima were defined by subsequent frequency calculations. Natural bond orbital analyses were performed using NBO 3.1 module of Gaussian 09 on optimized geometry.46–49 All calculated structures were visualized with ChemCraft.50
2.4 Electrochemistry of Cu(II)–(LH2)2
Cyclic voltammetry was performed on an EG & G Potentiostat Model 263A, Princeton Applied Research with power suite software for electrochemistry. Voltammograms were recorded using the three-electrode system. A glassy-carbon electrode of surface area 0.1256 cm2 was the working electrode, Ag/AgCl, KCl (saturated) was the reference while a platinum wire served as the counter electrode. Electrochemical measurements were undertaken in a 50 ml electrochemical cell with dimethyl formamide (DMF) as the solvent and 0.1 (M) tetrabutylammonium bromide (TBAB) as the supporting electrolyte.23,24 Before each cyclic voltammetry experiment, the solution was purged using high purity Argon for 40 minutes, while in between each scan the solution was purged with Argon for a further 5 minutes.
2.5 Interaction of LH3 and Cu(II)–(LH2)2 with CT DNA
2.5.1 By UV-Vis spectroscopy. Interaction was studied using a JASCO V-630 UV-Vis spectrophotometer. A pair of quartz cuvettes (STARNA Scientific Ltd.; 10 mm × 10 mm path length) was used. Separate aliquots, each containing a constant concentration (75 μM) of LH3 or Cu(II)–(LH2)2 and different concentrations of CT DNA were used at different pH. The concentration of CT DNA was gradually increased until saturation was reached. The total volume was kept constant at 2.0 ml using 15 mM Tris buffer and 120 mM NaCl. At the end of the titration, CT DNA was ∼30 fold greater than the concentration of compounds used. Each experiment was repeated thrice. The binding constant and site size of the interaction was determined using standard equations.23,24,33,51,52
2.5.2 By fluorescence spectroscopy. The interaction was studied using a HITACHI S-7000 fluorescence spectrophotometer.23,24,32 Solutions were excited at 515 nm and emission was measured at 582 nm at 298 K. The complex (75 μM) was taken in a fluorescence cuvette (10 mm × 10 mm path length) and titrated with increasing amounts of CT DNA. The total volume (2.0 ml) was constant using 15 mM Tris buffer (pH 7.4) and 120 mM NaCl. Here too, at the point of saturation, CT DNA was ∼30 fold greater than the concentration of Cu(II)–(LH2)2. The experiment was repeated thrice. The binding constant and the site size of the interaction was evaluated.
2.6 DNA relaxation assays for topoisomerase I and topoisomerase II enzymes
Recombinant human DNA topoisomerase I and human DNA topoisomerase II enzymes were purchased from TopoGEN Inc (Port Orange, Florida, USA). DNA relaxation assays for recombinant human DNA topoisomerase I and topoisomerase II enzymes were performed in the presence and absence of the compounds by briefly incubating 100 fmol of supercoiled pBS SK (+) DNA with 50 fmol of the enzyme in the reaction buffer provided with the enzymes. Dimethyl sulphoxide (DMSO) concentration was maintained at 1% (vehicle control). Camptothecin (CPT) and doxorubicin (DOX) were purchased from Sigma (St. Louis, MO, US) to be used as positive control drugs that stabilize covalent topoisomerase I-DNA complexes and covalent topoisomerase II-DNA complexes, respectively. The reactions were incubated at 37 °C for 30 minutes, loaded on 1% agarose gel and subjected to electrophoresis at 20 volts overnight. After completion of electrophoresis, gels were stained with 0.5 μg ml−1 ethidium bromide and viewed by Gel Doc 2000 (BioRad) under UV illumination. Relaxation was assessed by monitoring the decreased electrophoretic mobility of relaxed topoisomers of pBS SK (+) DNA.
2.7 DNA cleavage assay for topoisomerase I and topoisomerase II enzymes
DNA cleavage assay for topoisomerase I and topoisomerase II was performed in the presence or absence of the compounds by briefly incubating 100 fmol of supercoiled pBS (SK+) DNA with 500 fmol of enzyme. For topoisomerase I, the reaction buffer contained 10 mM Tris-HCl (pH 7.5), 50 mM KCl, 5 mM MgCl2, 0.1 mM EDTA and 15 μg ml−1 BSA while for topoisomerase II the assay was done in a mixture of the two reaction buffers provided with the enzyme. All reactions were performed in the presence of 400 μM N-acetyl cysteine (NAC) (Sigma) and 100 μM ascorbic acid (AA) (Sigma). Reactions were incubated at 37 °C for 30 minutes and stopped with 0.5% SDS. Enzymes were digested by proteinase K treatment. DMSO concentration was maintained at 1% (vehicle control). CPT and DOX were used as positive control drugs that stabilize covalent topoisomerase I-DNA complexes and covalent topoisomerase II–DNA complexes, respectively. Reactions were loaded on 1% agarose gel with 0.5 μg ml−1 ethidium bromide. Electrophoresis at 80 volts was carried out for 3 hours. After completion of electrophoresis, gels were viewed by Gel Doc 2000 (BioRad) under UV illumination.
2.8 Cell culture and cell viability assay
MOLT-4 cells were cultured in a RPMI medium (GIBCO, Invitrogen, Carlsbad, CA, US), supplemented with 10% fetal bovine serum (GIBCO), antibiotic mixture (1×) PSN (GIBCO) and gentamicin reagent solution (GIBCO). Cells were incubated in a humidified CO2 incubator at 37 °C. Etoposide (ETO) was purchased from Sigma (St. Louis, MO, US) to be used as a positive control drug along with CPT and DOX. These were dissolved in DMSO. MOLT-4 cells were seeded in 96 well plates for 24 hours before drug treatment. After 24 hours, the cells were treated with LH3, Cu(II)–(LH2)2 and the control compounds. DMSO concentration was less than 0.5%. After treatment for 72 hours cell viability was checked by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Briefly, the cells were washed with 1× PBS and treated with MTT for 4 hours at 37 °C. The precipitates were dissolved in DMSO and the plates were analyzed on a Thermo MULTISKAN EX plate reader at 595 nm.
2.9 Immunoband depletion assay
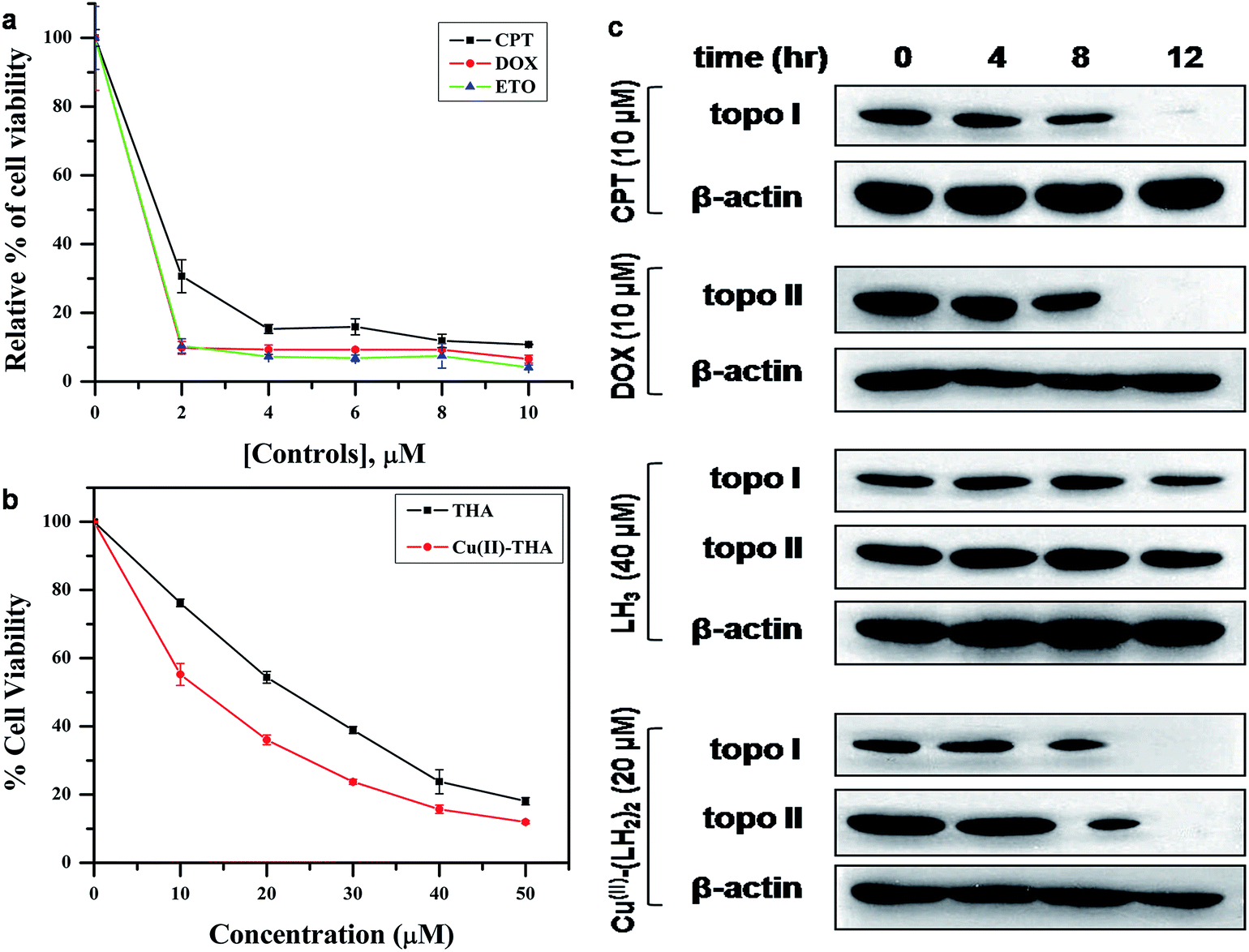
For the immunoband depletion assay MOLT-4 cells were cultured in 35 mm dishes and treated with either 10 μM CPT or 10 μM DOX or 40 μM LH3 or 20 μM Cu(II)–(LH2)2. Cells were harvested at 0 hours, 4 hours, 8 hours and 12 hours. Equal amounts of protein were subjected to electrophoresis on SDS–poly acryl amide gel. The separated proteins were then transferred onto a nitrocellulose membrane and western blotting was performed using anti-topo I and anti-topo II antibodies (Santa Cruz Biotechnology, Inc. Santa Cruz, CA, USA).
2.10 NADH dehydrogenase assay
The enzyme assay was undertaken at 298 K with cytochrome c as an electron acceptor.25,53 LH3 and Cu(II)–(LH2)2 were assayed for NADH-cytochrome c reductase activity, monitoring the reduction of cytochrome c at 550 nm. Tris buffer (pH ∼ 7.4), 80.0 μM cytochrome c, 160.0 μM NADH, 3.0 U per litre NADH dehydrogenase and test compounds were used. LH3 and Cu(II)–(LH2)2 concentrations were varied from 0 to 45.0 μM. The activity of NADH dehydrogenase is expressed in units, where one unit of activity reduces 1.0 μ mole oxidized cytochrome c per minute at 298 K. Formation of the superoxide radical anion catalyzed by LH3 and Cu(II)–(LH2)2 was measured from the reduction of cytochrome c inhibited by superoxide dismutase (SOD) (0 or 40.0 μg ml−1) in the presence of NADH and NADH dehydrogenase.25–27 The kinetics software of JASCO V-630 was utilized for the purpose.
3. Results and discussions
3.1 X-ray crystal structure description of [C28H14O10Cu]
Structural analysis from PXRD data (Fig. 1a) indicates that the Cu(II) complex crystallizes in the P space group. The ORTEP diagram for the complex is depicted in Fig. 1b. According to the refined structure obtained by analyzing the PXRD data, each asymmetric unit of the complex contains one Cu(II) ion, two monanionic LH2− units and two guest water molecules. Each Cu(II) center shows perfect square planar geometry with a coordination number of four. The coordination environment of Cu(II) is satisfied by two deprotonated phenolic –OH groups (O18 and O35) of two different LH3 units and two carbonyl oxygen atoms (O19 and O34). The crystal structure obtained by Rietveld refinement of the PXRD data shows that all Cu–O bond distances vary in the range 1.847–1.884 Å and all cisoid and transoid angles vary in the range 87.2–92.9° and 179.86–179.89°, respectively. Two LH3 units are inversely connected to the metal center; each monomeric unit is connected by supramolecular interactions leading to higher dimensions. The ORTEP diagram of the Cu(II)–(LH2)2 complex (Fig. 1b) almost completely matches the DFT optimized structure (Fig. 1c). In the absence of a single crystal for systems such as this one, powder X-ray diffraction remains the only option to obtain a structure. In our case, the structure obtained from powder diffraction not only matched other experimental findings but was also in good agreement with DFT calculations. This is the first reported structure of a complex of any hydroxy-9,10-anthraquinone with a 3d-transition metal ion.30 The final crystallographic data, structure refinement parameters, selected bond length and bond angles are summarized in Tables 1 and 2.
 |
| | Fig. 1 (a) The final Rietveld plot, where the red curve denotes the experimental pattern, the green curve denotes the simulated pattern and the pink curve indicates the difference of these two patterns. (b) ORTEP diagram of the complex Cu(II)–(LH2)2 (for clarity guest water molecules were omitted). Ellipsoids are drawn at 30% probability. (c) DFT optimized structure of Cu(II)–(LH2)2 complex. (d) to (g) area pictorial representation of respective MO diagrams of the Cu(II)–(LH2)2 complex. (h) Spin density plot of Cu(II)–(LH2)2 complex. | |
Table 1 Crystallographic data and refinement parameters obtained from PXRD data analysis
| Formula |
C28H10CuO12 |
| Formula weight |
601.92 |
| Crystal system |
Triclinic |
| Space group |
P |
| a/Å |
12.6644(14) |
| b/Å |
12.2799(10) |
| c/Å |
10.4602(13) |
| α/° |
97.155(11) |
| β/° |
113.937(9) |
| γ/° |
61.539(6) |
| V/Å3 |
1302.34(18) |
| Z |
2 |
| ρcalc/g cm−3 |
1.494 |
| Temperature/K |
293 |
| Radiation/Å |
1.54184 |
| 2θ range/° |
5–80 |
| Rwp |
0.0685 |
| Rp |
0.0429 |
Table 2 Selected bond lengths and bond angles of Cu(II)–(LH2)2 obtained by refinement of the PXRD pattern and those calculated by the DFT method
| Bonds |
Bond lengths (Å) |
| Obtained by refinement of the PXRD pattern |
Calculated by the DFT method |
| Cu1–O18 |
1.8469 |
1.919 |
| Cu1–O19 |
1.8839 |
1.954 |
| Cu1–O34 |
1.8843 |
1.954 |
| Cu1–O35 |
1.8467 |
1.919 |
|
| |
Bond angles (°) |
| O18–Cu1–O19 |
92.89 |
90.02 |
| O18–Cu1–O34 |
87.23 |
89.98 |
| O18–Cu1–O35 |
179.88 |
179.98 |
| O19–Cu1–O34 |
179.86 |
179.99 |
| O19–Cu1–O35 |
87.00 |
89.98 |
| O34–Cu1–O35 |
92.89 |
90.02 |
| Cu1–O18–C5 |
126.23 |
127.75 |
| Cu1–O19–C10 |
129.45 |
131.05 |
| Cu1–O34–C22 |
129.44 |
131.05 |
| Cu1–O35–C26 |
126.25 |
127.75 |
3.2 DFT study of Cu(II)–(LH2)2 complex
Although the structure of Cu(II)–(LH2)2 was obtained from powder X-ray diffraction data, a DFT study was carried out to check the formation of the complex and to predict certain physical and chemical properties. The MO composition of Cu(II)–(LH2)2 from DFT calculations is provided in Table 1S.† The α-HOMOs and β-LUMOs are primarily composed of redox non-innocent quinone moieties of Cu(II)–(LH2)2 that essentially make the paramagnetic copper centre a redox silent site (Fig. 1d–g). DFT calculations of Cu(II)–(LH2)2 predict quinone based redox processes. Analysis of the spin density plot (Fig. 1h) predicts a mixed metal–ligand behavior of the paramagnetic copper center, which was experimentally verified in the EPR spectrum of the complex (ESI†). The TDDFT calculation revealed that electronic transitions obtained in the UV-Vis spectrum would be LLCT (Table 3). DFT computer predictions of Cu(II)–(LH2)2 were in excellent agreement with the solved structure, with calculated bond lengths and angles being very close to those obtained through a computer model that provided a best fit for the experimental data (Fig. 1b, c, Tables 1 and 2).
Table 3 TDDFT table of the Cu(II)–(LH2)2 complex
| Energy (eV) |
Wavelength (nm) |
f |
Involved transition |
Character |
| 2.4741 |
501.13 |
0.3618 |
α HOMO−1 → α LUMO (0.57671) |
Ligand (pπ) → ligand (pπ*) |
| 3.2292 |
383.94 |
0.5022 |
β HOMO−6 → β-LUMO (0.70551) |
Ligand (pπ) → ligand (pπ*) |
3.3 Electrochemical behavior of Cu(II)–(LH2)2 complex
The complex showed one-electron reduction in DMF (Fig. 2a) with a peak at −0.866 V (vs. Ag,AgCl/KCl saturated) and E1/2 = −0.810 V was similar to an earlier report from our laboratory.32 In the case of LH3, we reported earlier that the molecule undergoes two-step one-electron reduction, forming a semiquinone that converts to a quinone di-anion.23 The variation of cathodic peak current with square root of potential sweep rate according to the Randles equation (eqn (1)) was linear in the case of the complex (Fig. 2b), indicating that the reduction of Cu(II)–(LH2)2 in DMF was diffusion controlled.55,56| | |
ipc = (2.69 × 105)n3/2D01/2ACν1/2
| (1) |
ipc = cathodic peak current (A), n = total number of electrons involved, A = area of the electrode (cm2), C = concentration (moles per cm3), ν = scan rate (V s−1). The diffusion coefficient (D0) was calculated from the slope of the plot (Fig. 2b) and found to be 4.462 × 10−5 cm2 s−1. The ratio of peak currents at different potential sweep rates were calculated with the help of Nicholson’s equation.56,57| |
 | (2) |
 |
| | Fig. 2 (a) Cyclic voltammogram of 120 μM of Cu(II)–(LH2)2 solution in DMF, recorded at 0.1 V s−1 potential sweep rate in 0.1 M TBAB as the supporting electrolyte, using the glassy carbon electrode (0.1256 cm2) at 298 K. (b) Linear dependence of cathodic peak current on the square root of the potential sweep rate for the one step two electron reduction of Cu(II)–(LH2)2 in DMF. | |
(isp)0 was the current at Eλ (switching potential) and (ipa)0 was the uncorrected anodic peak current with respect to zero current (baseline). The experimental data in Table 4 shows that the ratio of peak currents (ipa/ipc) was unity at all scan rates. This indicates that the redox behavior of Cu(II)–(LH2)2 in an aprotic solvent like DMF was completely reversible. Redox processes of the complex obtained from cyclic voltammetry studies corroborate the prediction made earlier from DFT calculations that redox processes would be quinone based. The Cu(II)/Cu(I) couple had a reduction potential of 0.55 V vs. Ag,AgCl/KCl (saturated) in an acetonitrile solvent.
Table 4 Electrochemical properties of single step one-electron reduction of Cu(II)–(LH2)2 in DMF
| ν (V s−1) |
ipa/ipc |
−E1/2 (V) |
| 0.025 |
1.0500 |
0.817 |
| 0.050 |
1.0175 |
0.812 |
| 0.075 |
1.0283 |
0.804 |
| 0.100 |
1.1208 |
0.801 |
| 0.200 |
1.0536 |
0.805 |
| 0.300 |
1.0339 |
0.819 |
| 0.400 |
1.0641 |
0.809 |
| 0.500 |
1.0768 |
0.820 |
| 0.750 |
1.0414 |
0.818 |
| 1.000 |
1.0233 |
0.826 |
3.4 Interaction of LH3 with CT DNA by UV-Vis spectroscopy: the effect of pH
A detailed study on the interaction of LH3 with CT DNA was reported earlier.23 In that report, we showed that LH3 is a DNA intercalator. Intrinsic binding constant and site size of the interaction was found to be (4.51 ± 0.20) × 104 M−1 and (5.21 ± 0.20) nucleotides, respectively.23 In this study, an effort was made to show the manner in which the intrinsic binding constant for the interaction of LH3 with DNA varied with pH at constant ionic strength of the medium. It was observed that as pH increased, the extent of interaction between the LH3 and CT DNA decreased (Table 2S†), attributed to the generation of an anionic form of LH3 at physiological pH.58 With increasing pH, LH2− formed from LH3 faced more repulsion from DNA, which was manifested in a decrease in binding constant values. Since LH3 exists in two forms at physiological pH [6.65 to 8.35], hence binding of it to CT DNA at any pH in the specified range was a consequence of both.58 To know the contribution of each and the impact that a change in pH has on the strength of interaction, the overall binding constant at each pH was treated according to the Henderson–Haselbach equation (eqn (3)).58,59| |
 | (3) |
The impact of the negative charge on LH2− on the overall binding constant was determined by plotting K* at five different pH values. Thus, the contribution of each form towards the overall binding constant was evaluated with the help of eqn (8) and (9). The overall binding constant could be defined as
| |
 | (4) |
The change in absorbance of LH3 upon binding enabled the determination of Cb and Cf (ESI†). [DNA] represents the total concentration of CT DNA. Since the overall binding constant at each pH was considered to be made up of two terms, K0 and K−, K0 was defined as the overall binding constant of the neutral form 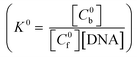 while K− was that due to the anionic form
while K− was that due to the anionic form  . The total bound and free forms of purpurin were
. The total bound and free forms of purpurin were
and
| |
 | (7) |
Eqn (4) could then be written as
| | |
K*(1 + 10pH–pK) = K0 + K− × 10pH–pK
| (8) |
or
| | |
K* = (K0 + K− × 10pH–pK)/(1 + 10pH–pK)
| (9) |
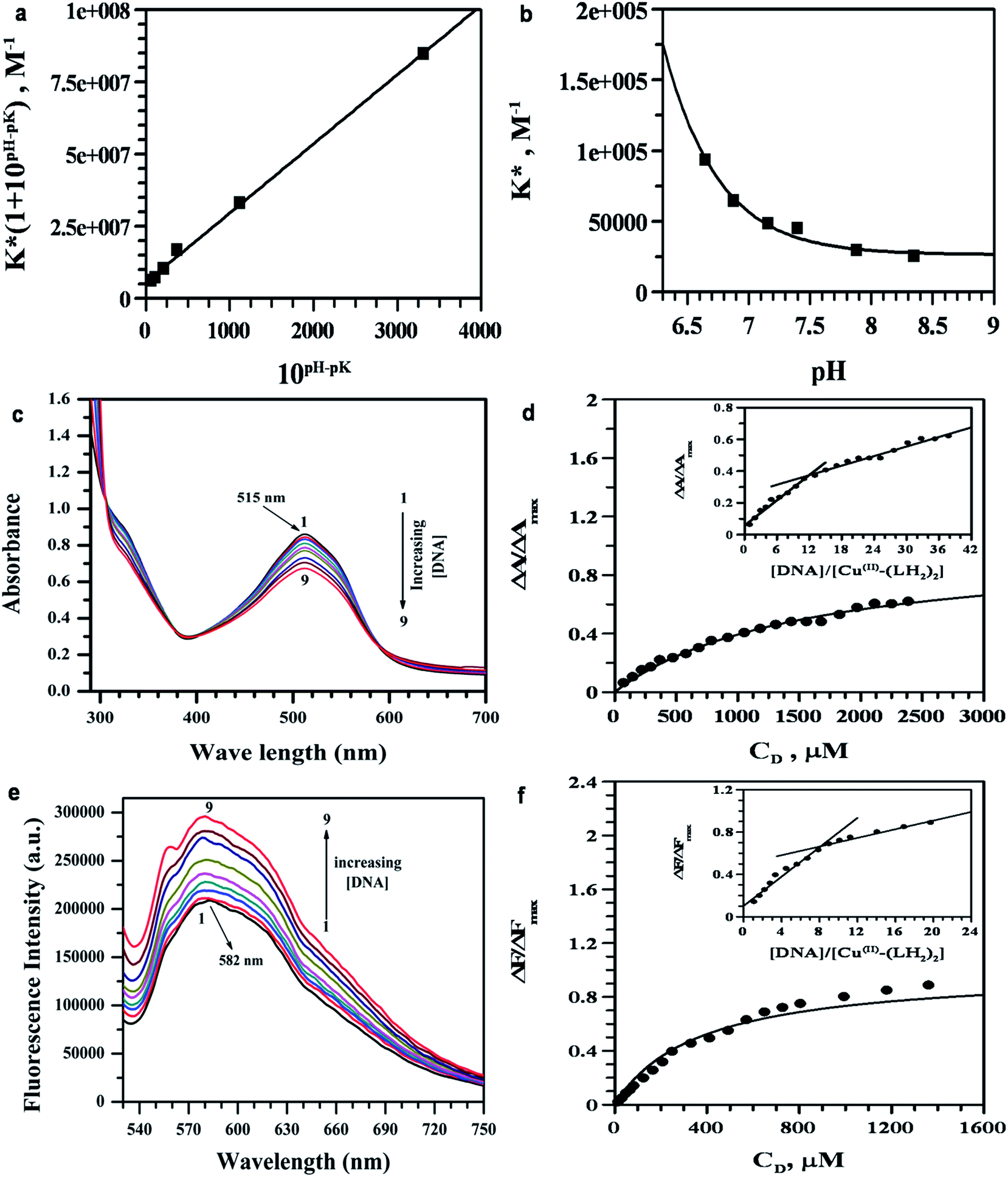
According to eqn (8), the plot of K*(1 + 10pH–pK) as a function of 10pH–pK yields a straight line (Fig. 3a) with correlation coefficient 0.99. K− was determined as the slope and K0 as the intercept. The values were (5.65 ± 0.48) × 106 M−1 and (2.41 ± 0.04) × 104 M−1 for K0 and K−, respectively. The overall binding constants (K*) were also plotted against pH according to eqn (9) (Fig. 3b) and values for K0 and K− were (4.61 ± 0.10) × 106 M−1 and (2.69 ± 0.07) × 104 M−1, respectively. Knowing the contributions of the neutral (K0) and mono-anionic (K−) forms of purpurin, one is now in a position to calculate K* for purpurin binding to CT DNA at any pH. The results indicate that the negative charge on the anionic form of LH3 (LH2−) holds the molecule back from interacting with CT DNA, which might have an impact on its overall potency.58,60 If the generation of the negative charge on LH3 is prevented then the overall binding constant (K*) should increase allowing purpurin to have binding constants with CT DNA that are comparable to those reported for anthracyclines.59 This is expected from a structure–function correlation that is today an important feature in understanding modern aspects in chemical biology.60
 |
| | Fig. 3 (a and b) Dependence of intrinsic binding constants (K*) for LH3 interacting with CT DNA for a variation of pH in the medium. An average of all K* values was considered for this plot. The solid line is the fitted data, which obeys eqn (8) [(a)] & eqn (9) [(b)]. [LH3] = 75 μM; [NaCl] = 120 mM; [Tris buffer] = 15 mM; T = 298 K. (c) Absorbance spectrum showing interaction of Cu(II)–(LH2)2 with CT DNA in the absence (1) and presence of (2) 296.83 μM, (3) 720.8729 μM, (4) 1218.027 μM, (5) 1682.03 μM, (6) 1974.56 μM, (7) 2523.05 μM, (8) 3027.66 μM, (9) 3493.46 μM CT DNA. (d) Binding isotherm for the spectrophotometric study of Cu(II)–(LH2)2 with CT DNA and the corresponding non-linear fit. Inset, the mole ratio plot of the same; (e) Fluorescence spectrum of Cu(II)–(LH2)2 interacting with CT DNA in the absence (1) and presence of (2) 15.50 μM, (3) 30.95 μM, (4) 77.14 μM, (5) 153.53 μM, (6) 304.04 μM, (7) 667.71 μM, (8) 1722.87 μM, (9) 2584.3 μM CT DNA. (f) Binding isotherm of fluorometric study of Cu(II)–(LH2)2 with CT DNA and the corresponding non-linear fit. Inset, the mole ratio plot of the same; [Cu(II)–(LH2)2] = 75 μM, [NaCl] = 120 mM; [Tris buffer] = 15 mM of pH 7.42; T = 298 K. | |
3.5 Interaction of Cu(II)–(LH2)2 with CT DNA
3.5.1 By UV-Vis spectroscopy. Cu(II)–(LH2)2 has a peak at 515 nm at physiological pH that gradually decreased in intensity on adding CT DNA. The spectra also show a slight blue shift by 4–7 nm (Fig. 3c). Interaction between the electronic states of the Cu(II)–(LH2)2 chromophore and those of the DNA bases could be a reason for this hypsochromic shift.23,24 These spectral features suggest that interaction between Cu(II)–(LH2)2 and CT DNA was a case of intercalation through an ordered stacking of Cu(II)–(LH2)2 between aromatic heterocyclic base pairs of the DNA helix.23,24,32 π–π stacking and dipole–dipole interactions help to stabilize the [Cu(II)–(LH2)2]–DNA adduct formed as a consequence of interaction between the electron-deficient quinones of Cu(II)–(LH2)2 and electron-rich purine or pyrimidine bases of the DNA helix.24The overall binding constant (K*) was determined with the help of standard equations (ESI†) [shown in Fig. 3d, S7(a) and (b)†]. The results are summarized in Table 5. Under conditions of 1:1 compound–DNA adduct formation, the presence of excess CT DNA compared to Cu(II)–(LH2)2, the Benesi–Hildebrand equation (eqn (10)) was used to determine K*.23,24,61
| |
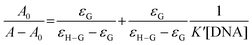 | (10) |
Table 5 Results of the binding parameters of Cu(II)–(LH2)2 with CT DNA by spectroscopic techniques
| Experiment |
Kapp × 10−3 (M−1) from double-reciprocal plot |
Kapp × 10−3 (M−1) from non-linear curve fitting |
K′ × 10−4 (M−1) = Kapp × nb |
K′ × 10−4 (M−1) Scatchard plot |
K′ × 10−4 (M−1) Benesi–Hildebrand double-reciprocal plot |
nb Scatchard plot |
| UV-Vis |
3.55 ± 0.42 |
1.72 ± 0.50 |
3.84 ± 0.15 |
4.80 ± 0.20 |
1.60 ± 0.20 |
10.82 ± 0.20 |
| Fluorimetry |
5.23 ± 0.22 |
6.28 ± 0.30 |
4.49 ± 0.28 |
4.04 ± 0.33 |
— |
8.58 ± 0.48 |
A0 and A are absorbances of Cu(II)–(LH2)2 in the absence and presence of CT DNA; εG and εH–G are absorption coefficients of Cu(II)–(LH2)2 and its adduct with DNA, respectively. The plot of A0/(A − A0) versus 1/[DNA] [Fig. S7(c)†] was linear. K* is reported in Table 5.
Upon complex formation, dissociation of the phenolic-OH at C2 of LH3 is retarded, with its pK value now being well beyond the physiological pH. As a result, formation of anionic species for Cu(II)–(LH2)2 did not arise and we found that, at all pH values (in the physiological pH range), the complex not only had a higher binding constant compared to purpurin (LH3), but more importantly the values remained constant over a considerable pH range.60 This aspect has a lot of significance for cancer patients for whom fluctuations in body pH occurs.
3.5.2 By fluorescence spectroscopy. Interaction of Cu(II)–(LH2)2 with CT DNA was followed at physiological pH by monitoring the increase in fluorescence upon adding CT DNA to a constant concentration of Cu(II)–(LH2)2. An increase in fluorescence was an indication that the mode of interaction was intercalation, as observed earlier for LH3.23 The emission spectrum of Cu(II)–(LH2)2 (Fig. 3e) exhibited a maximum at 582 nm, which was used to calculate the change in fluorescence (ΔF). Binding parameters for Cu(II)–(LH2)2 with CT DNA were analyzed using the same equations as mentioned for the UV-Vis study. For the purpose of analysis, the change in fluorescence intensity (ΔF) was considered instead of ΔA (equations are provided in the ESI†). In the case of fluorescence, our approach was based on the assumption that fluorescence intensity was linearly proportional to the concentration of Cu(II)–(LH2)2 bound to CT DNA. ΔFmax and Kapp (= Kd−1) were obtained from typical double reciprocal plots as the intercept and slope, respectively (Fig. S8(a)†). ΔF/ΔFmax was plotted against concentration of CT DNA. Applying non-linear fitting, Kapp (= Kd−1) was obtained (Fig. 3f). Data obtained from fluorimetric titration of Cu(II)–(LH2)2 with CT DNA was also analyzed according to Scatchard (Fig. S8(b)†).62 The results are summarized in Table 5. The data obtained from fluorescence and UV-Vis studies corroborate each other.
3.6 In vitro activity of LH3 and Cu(II)–(LH2)2 on human topoisomerase enzymes
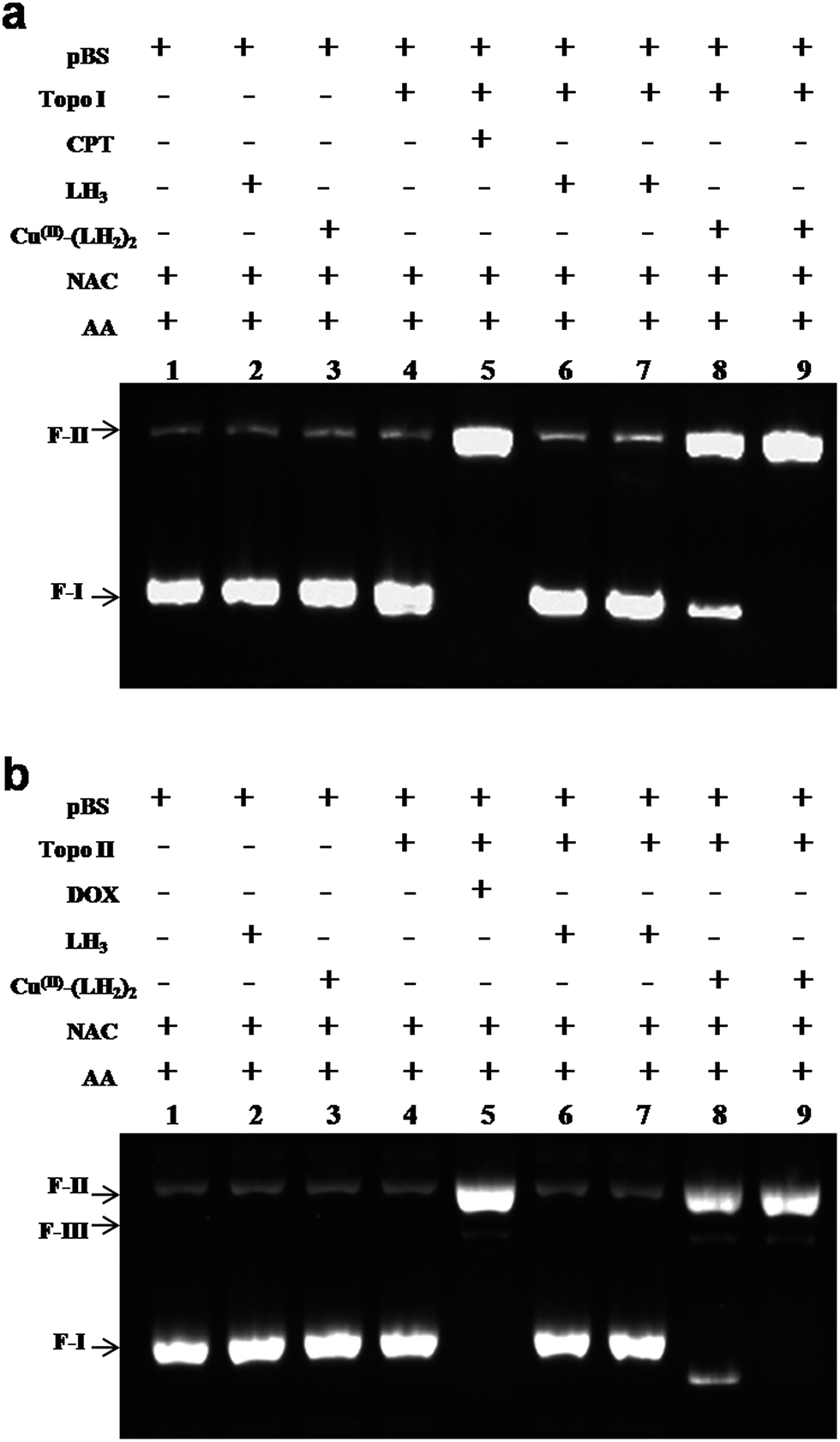
To check the inhibitory effects of LH3 and Cu(II)–(LH2)2 on human DNA topoisomerase I and human DNA topoisomerase II, DNA relaxation assays were performed using recombinant human DNA topoisomerase enzymes in the absence and presence of the compounds (Materials and Methods). The relaxation assay is based on the fact that supercoiled DNA molecules are relaxed by active topoisomerase enzymes, resulting in the formation of topoisomers of relaxed DNA molecules. These topoisomers migrate slowly compared to supercoiled plasmid DNA in agarose gel. Therefore, the activity of the enzymes may be checked by observing the gel bands for relaxed topoisomers. However, when the enzyme is inhibited, bands for the relaxed topoisomers do not appear on the gel. We found that Cu(II)–(LH2)2 completely inhibited DNA topoisomerase I and DNA topoisomerase II relaxation activities at a concentration of 20 μM, while LH3 had no effect at this concentration (Fig. 4a and b). We used established positive control inhibitors, CPT and DOX, for topoisomerase I and topoisomerase II enzymes, respectively. A kDNA decatenation assay was carried out for DNA topoisomerase II enzyme in the presence of LH3 and Cu(II)–(LH2)2. Cu(II)–(LH2)2 inhibited the decatenation activity of DNA topoisomerase II enzyme at 20 μM, while LH3 had no effect at this concentration (Fig. S9†). Therefore, our results suggest that Cu(II)–(LH2)2 is a novel potent dual inhibitor of topoisomerase I and topoisomerase II enzymes in vitro, similarly to CPT and DOX. Thus Cu(II)–(LH2)2 inhibit relaxation as well as decatenation activities of topoisomerase II enzymes.
 |
| | Fig. 4 DNA topoisomerase relaxation assays. (a) DNA topo I relaxation assay. Lane 1 is 100 fmol supercoiled pBS (SK+) DNA; lane 2 is 100 fmol supercoiled pBS (SK+) DNA with 40 μM LH3; lane 3 is 100 fmol supercoiled pBS (SK+) DNA with 40 μM Cu(II)–(LH2)2; lane 4 is 100 fmol supercoiled pBS (SK+) DNA with 50 fmol topoisomerase I enzyme; lane 5 is the same as lane 4 but with 10 μM CPT; lane 6 is the same as lane 4 but with 20 μM LH3; lane 7 is the same as lane 4 but with 40 μM LH3. Lane 8 is the same as lane 4 but with 10 μM Cu(II)–(LH2)2; lane 9 is the same as lane 4 but with 20 μM Cu(II)–(LH2)2. All the reactions were incubated at 37 °C for 30 minutes and analysed by agarose gel electrophoresis. (b) DNA topo II relaxation assay. Lane 1 is 100 fmol supercoiled pBS (SK+) DNA; lane 2 is 100 fmol supercoiled pBS (SK+) DNA with 40 μM LH3; lane 3 is 100 fmol supercoiled pBS (SK+) DNA with 40 μM Cu(II)–(LH2)2; lane 4 is 100 fmol supercoiled pBS (SK+) DNA with 50 fmol topoisomerase II enzyme; lane 5 is the same as lane 4 but with 10 μM DOX; lane 6 is the same as lane 4 but with 20 μM LH3; lane 7 is the same as lane 4 but with 40 μM LH3. Lane 8 is the same as lane 4 but with 10 μM Cu(II)–(LH2)2; lane 9 is the same as lane 4 but with 20 μM Cu(II)–(LH2)2. All the reactions were incubated at 37 °C for 30 minutes and analysed by agarose gel electrophoresis. | |
3.7 In vitro stabilization of covalent cleavage complexes by LH3 and Cu(II)–(LH2)2
In order to find out the type of topoisomerase inhibitor that the complex is, plasmid cleavage assays were performed for topoisomerase I and topoisomerase II enzymes, as described in Materials and Methods. A plasmid cleavage assay is based on the stabilization of covalent complexes between the enzyme and DNA, which decreases mobility of DNA in agarose gel. This is because during complex formation DNA is nicked, and nicked plasmid DNA has a lower electrophoretic mobility than supercoiled plasmid DNA. Therefore, when an inhibitor is of type class I it would stabilize covalent complexes that could be observed on agarose gel. The concentration of LH3 used was double (i.e. 40 μM) that of the complex [Cu(II)–(LH2)2] (20 μM), in order to confirm the fact that the effect was due to the complex as a whole and not due to the free ligand. Cu(II)–(LH2)2 stabilized the covalent enzyme–DNA complexes at 20 μM, while LH3 had no effect on such complex formation, even when used at 40 μM. CPT, which stabilizes covalent topoisomerase I-DNA complexes was used as a positive control (Fig. 5a). We further checked the effect of LH3 and Cu(II)–(LH2)2 on topoisomerase II-DNA covalent complex formation and found that Cu(II)–(LH2)2 completely stabilized the covalent topoisomerase II-DNA complex at 20 μM, while LH3 had no effect, even when used at 40 μM. DOX was used as the positive control drug, since it is known to stabilize covalent topoisomerase II-DNA complexes (Fig. 5b). Since Cu(II)–(LH2)2 generates ROS (although insignificant, discussed later), this could be responsible for DNA cleavage also. For this reason, cleavage assays were performed in the presence of NAC and AA, which are well-recognized ROS scavengers.63,64 With this result, one can now say that DNA cleavage was due to topoisomerase enzyme inactivation and not due to ROS generation (Fig. 5a and b). Together the results are interesting and allow us to speculate that Cu(II)–(LH2)2 could be a novel poison (class I inhibitor) for topoisomerase I and topoisomerase II enzymes.
 |
| | Fig. 5 Plasmid cleavage assays. (a) DNA topoisomerase I plasmid cleavage assay. All the cleavage assays were performed in the presence of NAC and ascorbic acid. Lane 1 is 100 fmol supercoiled pBS (SK+) DNA; lane 2 is 100 fmol supercoiled pBS (SK+) DNA with 40 μM LH3; lane 3 is 100 fmol supercoiled pBS (SK+) DNA with 40 μM Cu(II)–(LH2)2; lane 4 is 100 fmol supercoiled pBS (SK+) DNA with 500 fmol topoisomerase I enzyme; lane 5 is the same as lane 4 but with 10 μM CPT; lane 6 is the same as lane 4 but with 20 μM LH3; lane 7 is the same as lane 4 but with 40 μM LH3. Lane 8 is the same as lane 4 but with 10 μM Cu(II)–(LH2)2; lane 9 is the same as lane 4 but with 20 μM Cu(II)–(LH2)2. All the reactions were incubated at 37 °C for 30 minutes and stopped with 0.5% SDS. The enzyme was digested by proteinase K treatment and the reactions were analysed by agarose gel electrophoresis. (b) DNA topoisomerase II plasmid cleavage assay. Lane 1 is 100 fmol supercoiled pBS (SK+) DNA; lane 2 is 100 fmol supercoiled pBS (SK+) DNA with 40 μM LH3; lane 3 is 100 fmol supercoiled pBS (SK+) DNA with 40 μM Cu(II)–(LH2)2; lane 4 is 100 fmol supercoiled pBS (SK+) DNA with 500 fmol topoisomerase II enzyme; lane 5 is the same as lane 4 but with 10 μM DOX; lane 6 is the same as lane 4 but with 20 μM LH3; lane 7 is the same as lane 4 but with 40 μM LH3. Lane 8 is the same as lane 4 but with 10 μM Cu(II)–(LH2)2; lane 9 is the same as lane 4 but with 20 μM Cu(II)–(LH2)2. All the reactions were incubated at 37 °C for 30 minutes and stopped with 0.5% SDS. The enzyme was digested by proteinase K treatment and the reactions were analysed by agarose gel electrophoresis. | |
3.8 Effect of LH3 and Cu(II)–(LH2)2 on viability of MOLT-4 cells
Since DOX and other topo II inhibitors like etoposide (ETO) are used in the treatment of ALL we checked the effects of LH3 and Cu(II)–(LH2)2 on acute lymphoblastic leukemia (ALL) MOLT-4 cells. MTT assays were performed with CPT (IC50 = 8.89 ± 0.19 μM), ETO (IC50 = 5.59 ± 0.23 μM) and DOX (IC50 = 5.25 ± 0.55 μM) (Fig. 6a). The same cell viability assay was also carried out for LH3 and Cu(II)–(LH2)2 on ALL MOLT-4 cells. We found that Cu(II)–(LH2)2 kills MOLT-4 cells with an IC50 value of 18.59 ± 0.77 μM while the equivalent value for LH3 was 26.35 ± 0.31 μM, suggesting that Cu(II)–(LH2)2 was more potent than LH3 (Fig. 6b). We suspect that enhanced cell killing by Cu(II)–(LH2)2 was due to inhibition of the human DNA topoisomerase I and topoisomerase II enzymes present in the nucleus of MOLT-4 cells. Although IC50 for Cu(II)–(LH2)2 in MOLT-4 cells was higher compared to CPT, ETO and DOX, since Cu(II)–(LH2)2 produces relatively low ROS and exerts a novel dual inhibitory action on topoisomerase enzymes it could be developed as a useful and less costly alternative.
 |
| | Fig. 6 (a) Plot showing effects of CPT, ETO and DOX on MOLT-4 cells. (b) Dose response curve for effects of LH3 and Cu(II)–(LH2)2 on MOLT-4 cells. In both cases, MOLT-4 cells were treated with the respective compounds for 72 hours and a MTT assay was performed. (c) Immuno band depletion assay. Cultured MOLT-4 Cells were treated with the indicated concentrations of the compounds and harvested at 0 hour, 4 hours, 8 hours and 12 hours post treatment. CPT (10 μM) and DOX (10 μM) were used as positive controls. | |
3.9 Intracellular stabilization of topoisomerase–DNA covalent complexes by Cu(II)–(LH2)2
To check for intracellular stability of Cu(II)–(LH2)2 and formation of intracellular enzyme–DNA complexes we performed topoisomerase I and topoisomerase II immunoband depletion assay (IDA). IDA is based on the fact that if a topoisomerase inhibitor stabilizes the covalent enzyme–DNA complex inside the cell then enzyme molecules become associated with chromatin and will not be detected in the immunoband of the enzyme. In such a situation, the immunoband for topoisomerases will be depleted in a time dependent manner. Our results for topoisomerase I and topoisomerase II immunoband depletion assays (Fig. 6c) clearly showed that Cu(II)–(LH2)2 efficiently depletes the immunoband of both topoisomerase I and topoisomerase II after 12 hours of treatment with 20 μM Cu(II)–(LH2)2. On the contrary, LH3 did not deplete the immunoband after a similar 12 hour treatment with 40 μM (concentration of LH3 used was double that of Cu(II)–(LH2)2 in order to confirm that the effect was due to the complex and not the free ligand). These results indirectly indicate that Cu(II)–(LH2)2 is stable inside the cellular environment and shows without any doubt that it efficiently inhibits topoisomerase I and topoisomerase II enzymes by stabilizing covalent enzyme–DNA complexes.
3.10 NADH dehydrogenase assay of LH3 and Cu(II)–(LH2)2
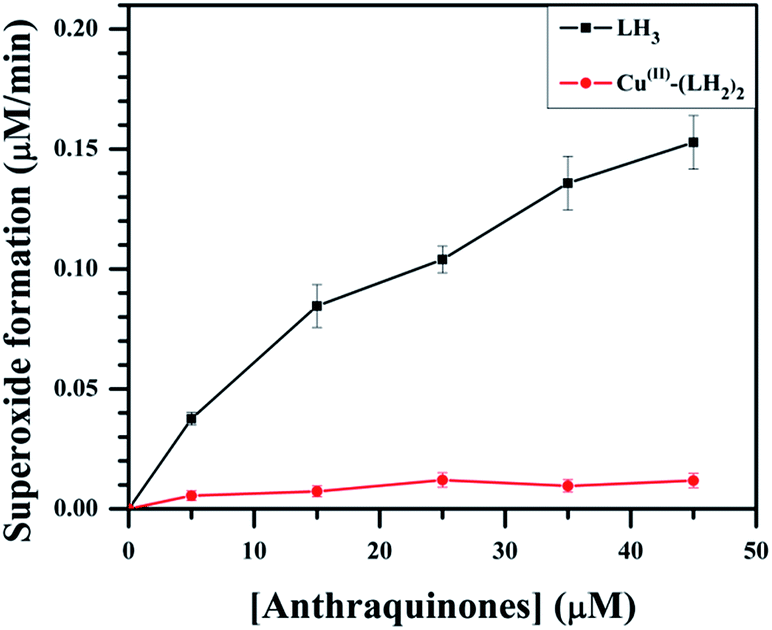
Several studies have suggested that the aspect of cardiotoxicity associated with anthracycline anticancer drugs is related to the formation of reduced oxygen species like the superoxide radical anion.1,10,11,16–18,26,27 It was shown earlier that metal complexes of anthracyclines generate less superoxide but are effective against tumors.27 Keeping this in mind, we compared the generation of superoxide by LH3 and Cu(II)–(LH2)2 by measuring the reduction of cytochrome c inhibited by superoxide dismutase (SOD).53 We found that on increasing the concentration of LH3, the yield of O2−˙ increased (Fig. 7), suggesting that LH3 catalyzed the flow of electrons from NADH to molecular oxygen through the enzyme NADH dehydrogenase. However, in the case of Cu(II)–(LH2)2, the formation of O2−˙ decreased considerably (Fig. 7). In these reactions, the formation of superoxide (O2−˙) occurs when semiquinones are oxidized by molecular oxygen.25–27 In the complex, the carbonyl at C9 of LH2− is involved in coordinating Cu(II), leading to a substantial decrease in semiquinone formation. Only one carbonyl (C10) on each LH2− is free to form semiquinones. Hence, much less semiquinone formation is expected for the complex. Moreover, owing to the presence of Cu(II) in the complex, the semiquinone formed quickly transfers its electron to the metal centre, leading to further decrease in its concentration.54 This affects its interaction with molecular oxygen, leading to decreased O2−˙ formation.
 |
| | Fig. 7 Effect of LH3 and Cu(II)–(LH2)2 on superoxide formation by NADH dehydrogenase, determined spectrophotometrically by the rate of SOD-inhibitable cytochrome-c reduction at pH 7.4 Tris buffer; [SOD] = 40 μg ml−1; [NADH] = 160 μM; [cytochrome c] = 80 μM; [NADH dehydrogenase] = 5 U per l. | |
4. Conclusions
A mononuclear complex of Cu(II) with LH3 [Cu(II)–(LH2)2] was prepared and characterized by physico-chemical and spectroscopic studies. Although a single crystal was not obtained, the structure was solved from X-ray powder diffraction data that was corroborated by a DFT study. Ours is the first reported crystal structure of any hydroxy-9,10-anthraquinone with a 3d-transition metal ion. Cyclic voltammetry of Cu(II)–(LH2)2 in DMF showed one-electron reduction [E1/2 = −0.810 V vs. Ag,AgCl/KCl (saturated)], which occurred at the quinone moiety of the ligand. The findings from cyclic voltammetry were corroborated by predictions from a DFT study that suggested redox processes would be quinone based. Physicochemical and electrochemical attributes of purpurin and its Cu(II) complex were similar to the anthracycline drug doxorubicin and its Cu(II) complex. The interaction of the complex with CT DNA suggests that, while in the case of purpurin the binding constant values decreased with increasing pH, complex formation could prevent this trend. For the complex, binding with CT DNA not only increased but remained constant over a wide range of pHs, improving the latter’s applicability. In vitro DNA topoiomerase relaxation assays showed that [Cu(II)–(LH2)2] was a dual poison for human DNA topoisomerase I and human DNA topoisomerase II enzymes, such as those known for CPT and DOX, which was a significant improvement following complex formation. DNA cleavage assays for topoisomerase I and topoisomerase II in the presence of the complex and known ROS scavengers revealed that DNA cleavage was due to the stabilization of the enzyme–DNA adduct and not due to ROS generation. With the help of ALL MOLT-4 cells, we could show that the complex inhibits topoisomerase I and topoisomerase II enzymes forming enzyme–DNA covalent complexes within the cells, as revealed by the results of the immunoband depletion assay. The NADH dehydrogenase assay performed for determining ROS generation by the compounds revealed that the generation of superoxide radicals by Cu(II)–(LH2)2 was much lower than LH3.
Since the complex was found to be more potent in killing ALL MOLT-4 cells than LH3 and considering findings on DNA topoisomerase enzymes, it may be suggested that the complex targets topoisomerase I and topoisomerase II enzymes during anticancer activity. Findings from this study illustrate that Cu(II)–(LH2)2 is a promising anticancer agent. Further understanding of the mode of action of the complex may rationally help to modify the chemical and biological properties to optimize anticancer activity.
Abbreviations
| LH3 | Purpurin or 1,2,4-trihydroxy-9,10-anthraquinone |
| Cu(II)–(LH2)2 | Cu(II) complex of purpurin or LH3 |
| CT DNA | Calf thymus DNA |
| ROS | Reactive oxygen species |
| SOD | Superoxide dismutase |
| ETO | Etoposide |
| CPT | Camptothecin |
| DOX | Doxorubicin |
| NAC | N-Acetyl acetic acid |
| AA | Ascorbic acid |
Acknowledgements
Financial support from DST, Govt. of West Bengal [794(Sanc.)1(10) ST/P/S&T/9G-23/2013] in the form of a research project to S. D. is gratefully acknowledged. P. D. expresses his gratitude to the University Grants Commision, Government of India for a research fellowship. C. K. J. also acknowledges the University Grants Commision, Government of India, for providing a research fellowship. S. D. offers his gratitude to Prof. Samita Basu, Head, CSD, SINP and former Head, Prof. Soumen Basak for allowing P. D. to carry out experiments on fluorescence and cyclic voltammetry. S. D. also wishes to thank Prof. Amitabha De and his student Ankan Dutta Chowdhury of CSD, SINP for their co-operation in carrying out cyclic voltammetry experiments.
References
- G. Minotti, P. Menna, E. Salvatorelli, G. Cairo and L. Gianni, Pharmacol. Rev., 2004, 56, 185–229 CrossRef CAS PubMed.
- K. C. Chow, T. L. Macdonald and W. E. Ross, Mol. Pharmacol., 1988, 34, 467–473 CAS.
- J. C. Wang, Nat. Rev. Mol. Cell Biol., 2002, 3, 430–440 CrossRef CAS PubMed.
- D. A. Koster, A. Crut, S. Shuman, M. Bjornsti and N. H. Dekker, Cell, 2010, 142, 519–530 CrossRef CAS PubMed.
- Y. Pommier, Chem. Rev., 2009, 109, 2894–2902 CrossRef CAS PubMed.
- Y. Pommier, Curr. Med. Chem.: Anti-Cancer Agents, 2004, 5, 429–434 CrossRef.
- Y. Pommier, E. Leo, H. L. Zhang and C. Marchand, Chem. Biol., 2010, 17, 421–433 CrossRef CAS PubMed.
- Y. Pommier, Nat. Rev. Cancer, 2006, 6, 789–802 CrossRef CAS PubMed.
- F. Arcamone and S. Penco, in Anthracycline and Anthracenedione-Based Anticancer Agents, ed. J. W. Lown, Elsevier, Amsterdam, 1988 Search PubMed.
- A. J. M. Ferreri, E. Campo, A. Ambrosetti, F. Ilariucci, J. F. Seymour, R. Willemze, G. Arrigoni, G. Rossi, A. Lopez-Guillermo, E. Berti, M. Eriksson, M. Federico, S. Cortelazzo, S. Govi, N. Frungillo, S. Dell’Oro, M. Lestani, S. Asioli, E. Pedrinis, M. Ungari, T. Motta, R. Rossi, T. Artusi, P. Iuzzolino, E. Zucca, F. Cavalli and M. Ponzoni, Ann. Oncol., 2004, 15, 1215–1221 CrossRef CAS PubMed.
- E. V. Barry, S. E. Lipshultz and S. E. Sallan, Anthracycline-induced cardiotoxicity: natural history, risk factors, and prevention, in American Society of Clinical Oncology 2008 Educational Book, ed. R. Govindan, American Society of Clinical Oncology, Alexandria, 2008, pp. 448–453 Search PubMed.
- K. Schimmel, D. Richel, R. van den Brink and H. J. Guchelaar, Cancer Treat. Rev., 2004, 30, 181–191 CrossRef CAS PubMed.
- M. I. Gharib and A. K. Burnett, Eur. J. Heart Failure, 2002, 4, 235–242 CrossRef CAS.
- L. C. M. Kremer, E. C. V. Dalen, M. Offringa and P. A. Voute, Ann. Oncol., 2002, 13, 503–512 CrossRef CAS PubMed.
- S. M. Swain, F. S. Whaley and M. S. Ewer, Cancer, 2003, 97, 2869–2879 CrossRef CAS PubMed.
- D. Iarussi, P. Indolfi, F. Casale, V. Martino, M. T. Di Tullio and R. Calabrò, Pediatr. Drugs, 2005, 7, 67–76 CrossRef PubMed.
- D. L. Keefe, Semin. Oncol., 2001, 28, 2–7 CrossRef CAS PubMed.
- D. Cardinale, A. Colombo, G. Lamantia, N. Colombo, M. Civelli, G. D. Giacomi, M. Rubino, F. Veglia, C. Fiorentini and C. M. Cipolla, J. Am. Coll. Cardiol., 2010, 55, 213–220 CrossRef CAS PubMed.
- J. L. Speyer, M. D. Green, A. Zeleniuch-Jacquotte, J. C. Wernz, M. Rey, J. Sanger, E. Kramer, V. Ferrans, H. Hochster and M. Meyers, J. Clin. Oncol., 1992, 10, 117–127 CAS.
- A. Moreno-Aspitia and E. A. Perez, Clin. Ther., 2009, 31, 1619–1640 CrossRef CAS PubMed.
- T. Pecere, M. V. Gazzola, C. Mucignat, C. Parolin, F. D. Vecchia, A. Cavaggioni, G. Basso, A. Diaspro, B. Salvato, M. Carli and G. Palù, Cancer Res., 2000, 60, 2800–2804 CAS.
- G. Cozza, M. Mazzorana, E. Papinutto, J. Bain, M. Elliott, G. di Maira, A. Gianoncelli, M. A. Pagano, S. Sarno, M. Ruzzene, R. Battistutta, F. Meggio, S. Moro, G. Zagotto and L. A. Pinna, Biochem. J., 2009, 421, 387–395 CrossRef CAS PubMed.
- P. Das, P. S. Guin, P. C. Mandal, M. Paul, S. Paul and S. Das, J. Phy. Org. Chem., 2011, 24, 774–785 CAS.
- P. S. Guin, S. Das and P. C. Mandal, J. Phys. Org. Chem., 2010, 23, 477–482 CrossRef CAS.
- S. Das, A. Saha and P. C. Mandal, Talanta, 1996, 43, 95–102 CrossRef CAS.
- M. M. L. Fialo and A. G. Suillerot, Biochim. Biophys. Acta, Gen. Subj., 1985, 840, 91–98 CrossRef.
- H. Beraldo, A. Garnier-Suillerot, L. Tosi and F. Lavelle, Biochemistry, 1985, 24, 284–289 CrossRef CAS.
- C. Santini, M. Pellei, V. Gandin, M. Porchia, F. Tisato and C. Marzano, Chem. Rev., 2014, 114, 815–862 CrossRef CAS PubMed.
- A. Kheirolomoom, L. M. Mahakian, C. Y. Lai, H. A. Lindfors, J. W. Seo, E. E. Paoli, K. D. Watson, E. M. Haynam, E. S. Ingham, L. Xing, R. H. Cheng, A. D. Borowsky, R. D. Cardiff and K. W. Ferrara, Mol. Pharmaceutics, 2010, 7, 1948–1958 CrossRef CAS PubMed.
- M. D. Vaira, P. Orioli, F. Piccioli, B. Bruni and L. Messori, Inorg. Chem., 2003, 42, 3157–3159 CrossRef PubMed.
- D. Cova, M. Sassano, E. Monti and F. Piccinini, Arch. Toxicol., 1990, 64, 597–598 CrossRef CAS.
- P. S. Guin, S. Das and P. C. Mandal, J. Inorg. Biochem., 2009, 103, 1702–1710 CrossRef CAS PubMed.
- Solvent extraction in Textbook of Quantitative Chemical analysis, ed. G. H. Jeffery, J. Bassett, J. Mendham and R. C. Denney, 5th edn, ELBS, Longman, Great Britain, 1989, pp. 178–184 Search PubMed.
- A. Altomare, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, R. Rizzi and P. E. Werner, J. Appl. Crystallogr., 2000, 33, 1180–1186 CrossRef CAS.
- V. Favre-Nicolin and R. Cerny, J. Appl. Crystallogr., 2002, 35, 734 CrossRef CAS . http://objcryst.sourceforge.net.
- J. J. P. Stewart, MOPAC 5.0, A general purpose Molecular Orbital Package (QCEP 455) Search PubMed.
- A. C. Larson and R. B. Von Dreele, General Structure Analysis System (GSAS), Los Alamos National Laboratory Report LAUR (2000) 86–748.
- B. H. Toby, J. Appl. Crystallogr., 2001, 34, 210–213 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 Revision A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- T. H. Dunning Jr and P. J. Hay, in Modern Theoretical Chemistry, ed. H. F. Schaefer, New York, 1976, vol. III, pp. 1–28 Search PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS PubMed.
- N. M. O’Boyle, GaussSum 2.1, 2007, available at http://www.gausssum.sf.net Search PubMed.
- N. M. O’Boyle, A. L. Tenderhol and K. M. Langner, J. Comput. Chem., 2008, 29, 839–845 CrossRef PubMed.
- E. D. Glendening, A. E. Reed, J. E. Carpenter and F. Weinhold, NBO version 3.1, Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 2001 Search PubMed.
- A. E. Reed and F. Weinhold, J. Chem. Phys., 1985, 83, 1736–1740 CrossRef CAS PubMed.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- F. Weinhold, in Natural Bond Orbital Methods in Encyclopedia of Computational Chemistry, ed. P. V. R. Schleyer, Wiley, UK, 1998, pp. 1792–1811 Search PubMed.
- D. A. Zhurko and G. A. Zhurko, ChemCraft 1.5, Plimus, San Diego, CA, p. 92130, http://www.chemcraftprog.com Search PubMed.
- S. Roy, R. Banerjee and M. Sarkar, J. Inorg. Biochem., 2006, 100, 1320–1331 CrossRef CAS PubMed.
- S. Chakraborti, B. Bhattacharyya and D. Dasgupta, J. Phys. Chem. B, 2002, 106, 6947–6953 CrossRef.
- H. R. Mahler, in Methods in Enzymology 11, ed. S. P. Colowick and N. O. Kaplan, Academic Press, New York, 1955, pp. 668–672 Search PubMed.
- S. Das, A. Bhattacharya, P. C. Mandal, M. C. Rath and T. Mukherjee, Radiat. Phys. Chem., 2002, 65, 93–100 CrossRef CAS.
- J. E. B. Randles, Trans. Faraday Soc., 1948, 44, 322–327 RSC.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods Fundamental and Applications, John Wiley & Sons, New York, 2nd edn, 2001 Search PubMed.
- R. S. Nicholson, Anal. Chem., 1966, 38, 1406 CrossRef CAS.
- S. Mukherjee, P. Das and S. Das, J. Phys. Org. Chem., 2012, 25, 385–393 CrossRef CAS.
- F. Frezard and G. Suillerot, Biochim. Biophys. Acta, Gen. Subj., 1990, 1036, 121–127 CrossRef CAS.
- S. Mukherjee, P. Gopal, S. Paul and S. Das, J. Anal. Oncol., 2014, 3 Search PubMed.
- H. A. Benesi and J. H. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703–2707 CrossRef CAS.
- G. Scatchard, Ann. N. Y. Acad. Sci., 1949, 51, 660–672 CrossRef CAS PubMed.
- S. M. Attia, Arch. Toxicol., 2012, 86, 725–731 CrossRef CAS PubMed.
- S. Belin, F. Kaya, G. Duisit, S. Giacometti, J. Ciccolini and M. Fontés, PLoS One, 2009, 4(2), e4409, DOI:10.1371/journal.pone.0004409.
Footnote |
| † Electronic supplementary information (ESI) available: Physicochemical experiments on complex formation in solution. Spectroscopic characterization (IR, Mass, EPR) of the complex. Table showing TDDFT calculation. Various plots for evaluation of binding parameters of LH3 and Cu(II)–(LH2)2 with CT DNA. CIF files of Cu(II)–(LH2)2. CCDC 916875. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra07127a |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here. 






 while K− was that due to the anionic form
while K− was that due to the anionic form 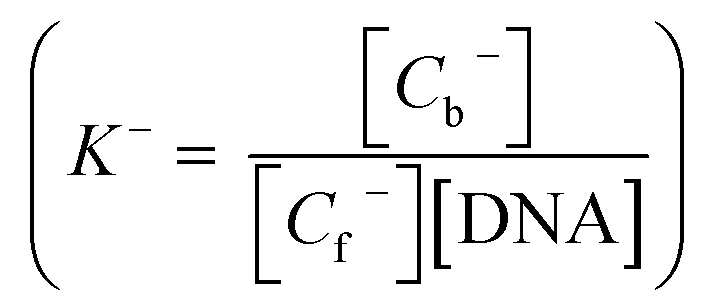 . The total bound and free forms of purpurin were
. The total bound and free forms of purpurin were