
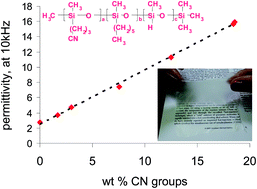
Abstract
New polymers with tuneable dielectric properties were prepared by modifying trimethylsilyl end-terminated poly(methylhydro)siloxane with polar γ-cyanopropyl groups. The amount of polar groups was tuned by adjusting the allyl cyanide/n-hexene ratio in poly(methylhydro)siloxane co-hydrosilylation. The copolymers were characterized by FTIR and NMR spectroscopy. The distribution of the polar groups along the chain was evaluated based on 1H NMR spectroscopy. The influence of the amount of polar γ-cyanopropyl on the glass transition temperature (Tg) and on the dielectric properties was investigated by DSC and impedance spectrometry. All polymers showed Tgs well below room temperature. A linear increase in permittivity (ε′) with increasing amount of γ-cyanopropyl groups was observed. A maximum ε′ value of 15.9 for the copolymer containing 89 mol% polar groups was achieved, which is 6-fold higher than polydimethylsiloxane. The incomplete conversion of Si–H groups observed in all hydrosilylation reactions with allyl cyanide opened up the possibility of using the prepared copolymers as cross-linkers.
 Please wait while we load your content...
Please wait while we load your content...

