
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceKinetics of hydrodeoxygenation of octanol over supported nickel catalysts: a mechanistic study†
Venkata Chandra Sekhar Palla ,
Debaprasad Shee* and
Sunil K. Maity
,
Debaprasad Shee* and
Sunil K. Maity
Department of Chemical Engineering, Indian Institute of Technology Hyderabad, Ordnance Factory Estate, Yeddumailaram 502205, Medak, Telengana, India. E-mail: dshee@iith.ac.in; Fax: +91 40 2301 6003; Tel: +91 40 2301 6109
First published on 28th August 2014
Abstract
The hydrodeoxygenation (HDO) of 1-octanol as a model aliphatic alcohol of bio-oil was investigated in a continuous down-flow fixed-bed reactor over γ-Al2O3, SiO2, and HZSM-5 supported nickel catalysts in the temperature range of 488–533 K. The supported nickel catalysts were prepared by incipient wetness impregnation method and characterized by BET, XRD, TPR, TPD, H2 pulse chemisorption, and UV-vis spectroscopy. Characterization of supported nickel (or nickel oxide) catalysts revealed existence of dispersed as well as bulk nickel (or nickel oxide) depending on the extent of nickel loading and the nature of the support. The acidity of γ-Al2O3 supported nickel catalysts decreased with increasing the nickel loading on γ-Al2O3. n-Heptane, n-octane, di-n-octyl ether, 1-octanal, isomers of heptene and octene, tetradecane, and hexadecane were identified as products of HDO of 1-octanol. The C7 hydrocarbons were observed as primary products for catalysts with active metal sites such as γ-Al2O3 and SiO2 supported nickel catalysts. However, C8 hydrocarbons were primarily formed over acidic catalysts such as pure HZSM-5 and HZSM-5 supported nickel catalyst. The 1-octanol conversion increased with increasing nickel loading on γ-Al2O3, and temperature and decreasing pressure and WHSV. The selectivity to products was strongly influenced by temperature, nickel loading on γ-Al2O3, pressure, and types of carrier gases (nitrogen and hydrogen). The selectivity to C7 hydrocarbons was favoured over catalysts with increased nickel loading on γ-Al2O3 at elevated temperature and lower pressure. A comprehensive reaction mechanism of HDO of 1-octanol was delineated based on product distribution under various process conditions over different catalysts.
1 Introduction
At present our society is heavily dependent on finite fossil fuels to meet the growing demands of energy and organic chemicals. Currently, more than 80% of energy and about 90% of organic chemicals of the world are derived from fossil fuels alone.1 The continuous decline of fossil fuels reserves, increase of fuel prices, and deterioration of environmental cleanliness due to large scale usage of fossil fuels have stimulated intense research worldwide to pursue carbon neutral renewable sources of energy and organic chemicals to reduce reliance on limited fossil fuels. Several renewable energies including hydro, solar, wind, geothermal, and biomass were thus developed in the past to alleviate the fossil fuel crisis. However, with the exception of biomass, all these renewable energies can only fulfil the global energy demands to some extent in the form of heat and electricity only. On the other hand, biomass, being the origin of fossil fuels, has enormous potential to deliver all forms of energy (heat, electricity, and transportation fuels) as well as basic organic chemicals in an integrated biorefinery.Several biomass conversion technologies including gasification, fast pyrolysis, transesterification, and fermentation have thus been progressed considerably to fulfil goals of integrated biorefinery. The fast pyrolysis is one such promising technology for thermochemical conversion of world's most abundant lignocellulosic biomass directly into liquid products commonly known as bio-oil.2,3 Low capital investments and economic viability at small scale make fast pyrolysis an attractive choice for decentralized biorefinery. The bio-oil, being a mixture of more than 300 identified oxygenated hydrocarbons, could be considered as a potential feedstock for basic organic chemicals as well as transportation fuels.4 However, separation of the compounds of very low concentration from mixtures of large number of chemical compounds of many classes is practically impossible and hence bio-oil is inappropriate as feedstock for organic chemicals. The bio-oil is also unsuitable for direct usage as transportation fuels in internal combustion engines because of its high water and oxygen contents, immiscibility with petroleum fuels, low heating value (∼40–45% of hydrocarbon fuels), poor storage stability due to unsaturated compounds, and high corrosiveness due to organic acids mainly acetic and formic acid.5–7 The removal of oxygen from bio-oils is thus indispensable for production of liquid transportation fuels.
The hydrodeoxygenation (HDO) is a propitious technology for removal of oxygen from bio-oils thereby increasing volatility and thermal stability and reducing viscosity.8 Knowledge of reaction mechanism plays crucial role for design of suitable catalysts for cost-effective HDO of bio-oils. Understanding mechanism of HDO of whole bio-oils is however highly challenging because of its diverse functionalities (phenolics, aldehydes, ketones, alcohols, organic acids, and others). The present work was therefore commenced to explore fundamental understanding of HDO of 1-octanol as a model aliphatic alcoholic functionality of bio-oil.
Enormous research efforts have been made in the past on mechanistic understanding of HDO of whole bio-oil2,3 and various oxygenated compounds of bio-oil such as phenol, anisole, guaiacol,9–13 aliphatic aldehydes like heptanal,14–16 aromatic aldehydes like furfural,17,18 benzaldehyde, 2-phenylpropionaldehyde, cinnamaldehyde, 3-phenylpropionaldehyde, and 4-isopropylbenzaldehyde,16 and various ketones such as 6-undecanone, 2-dodecanone, 2,4-dimethyl-3-petnanone, 2,2,4,4-tetramethyl-3-pentanone,14 cyclopentanone,19 and methyl isobutyl ketone.20 However, limited information is available in open literatures on HDO of aliphatic alcohols.14,21–24 The HDO of 1-heptanol was first reported over commercial sulphided NiMo/γ-Al2O3 and CoMo/γ-Al2O3 catalysts in both flow (at 523 K and 15 bars of H2) and batch (at 523 K and 75 bars of H2 or N2) reactor using m-xylene or n-dodecane as solvent.21,22 The work was further extended in a flow reactor using two different H2S concentration in the feed.23 Heptane and heptenes together with small quantity of diheptyl ether were observed as products.21–23 Donnis et al. reported HDO of 3-heptanol in a microscale reactor over commercial sulfided NiMo catalyst at 453 and 483 K and 50 bars of H2 using n-octane as solvent.13 Very recently, Peng et al. reported HDO of aqueous 1- and 2-propanol in a batch reactor in presence of 3 wt% Pt/Al2O3 at 473 K and 40 bars of H2.23 Propane, ethane, CO2, and acetone (in case of 2-propanol only) were observed as products. The HDO of 2-propanol was also carried out in a fixed-bed reactor under vapor phase at different temperatures (393–523 K) using hydrogen as carrier gas.
It is quite evident from above discussion that detailed study on HDO of neat aliphatic alcohols was not attempted in the past over inexpensive nickel based catalysts to establish a comprehensive reaction mechanism. A systematic investigation was therefore commenced in the present work on HDO of 1-octanol in a fixed-bed reactor under various process conditions over several supported (γ-Al2O3, SiO2, and HZSM-5) nickel catalysts with different degree of acidity to delineate a holistic reaction mechanism.
2 Experimental
2.1 Catalyst preparation
γ-Al2O3 (Alfa Aesar, 99.9% purity), SiO2 (Aerosil-200), and HZSM-5 (Zeolyst) supported nickel catalysts were prepared by incipient wetness impregnation method using nickel nitrate hexahydrate (Ni(NO3)2·6H2O, Merck, >97% purity) as precursor. The nickel precursor solution was first prepared by dissolving measured amount of precursor corresponding to desired nickel loading in distilled water of volume equal to or slight excess of total pore volume of the support. The precursor solution was then added to the support and resultant mixture was agitated continuously for 2 h for uniform distribution of metal precursor solution into the support. The wet material was then dried overnight at 353 K for 12 h. The dried material was finally calcined at 823 K for 6 h. The supported nickel oxide catalysts obtained after calcination was referred as oxidized catalyst throughout the article. The γ-Al2O3 supported nickel catalysts were designated as xNiAl (x = 5–20 wt% nickel). The HZSM-5 (Zeolyst) with Si/Al ratio of 23 and 55 were designated as ZSM23 and ZSM55 respectively. The 15 wt% nickel supported on SiO2, ZSM23, and ZSM55 were represented as 15NiSi, 15NiZSM23, and 15NiZSM55 respectively.2.2 Catalyst characterization
2.3 Experimental setup and procedure
The HDO of 1-octanol was performed in an isothermally operated continuous down-flow fixed-bed reactor using either nitrogen or hydrogen as carrier gas. The known amount of oxidized catalyst diluted with appropriate quantity of quartz beads was placed inside a stainless steel tubular reactor (1/2′′ O.D. and 40 cm length) reinforced by quartz wool from either side.25 The reactor was then kept inside a tubular furnace consisting of two heating zones (preheating and reaction). The temperature of the reaction zone was controlled by a PID controller within ±1 K. The flow rate of all gases was controlled by individual mass flow controllers (Bronkhorst, EL-Flow). The oxidized catalysts were first reduced by hydrogen (25 mL min−1) at 923 K for 3 h. The supported nickel catalysts obtained after reduction were referred as reduced catalyst throughout the article. The reactor was then cooled down to desired reaction temperature and 1-octanol was pumped into the reactor at specified flow rate using a HPLC pump (Lab alliance, Series-I). The product gases leaving the reactor were then passed through a vertical condenser. The non-condensable gases separated in a gas-liquid separator were subsequently analyzed online by gas chromatograph (GC) equipped with TCD detector and Carbosieve-S-II packed column (Chromatopak, 3 m × 1/8′′) using argon as carrier gas. Liquid samples were collected at regular interval of time after reaching steady state condition. The products were identified using GC attached with mass spectroscopy (Shimadzu, GCMS-QP2010 Ultra). Occurrence of the products was confirmed by comparing standard mass fragment data available with data acquisition software (Wiley and NIST standard libraries). The products were quantified by GC (Shimadzu, GC-2014) using capillary column (ZB-5HT Inferno, Phenomenex, 30 m × 0.32 mm × 0.10 μm) connected with flame ionization detector (FID). The following GC oven temperature programming was used to obtain well resolved chromatogram: Initially oven was kept at 313 K for 3 minutes and then temperature was increased to 493 K with a ramping rate of 15 K min−1, and held at this temperature for 3 min. Injector and detector were maintained at 493 K and 523 K respectively. The material balance was checked for all experimental runs and errors were within ±3%.3 Results and discussion
3.1 Catalyst characterization
The BET surface area and pore volume of the oxidized catalysts and pure supports are shown in Table 1. The surface area of the oxidized catalysts was somewhat lesser than pure supports. The decrease of surface area might be due to blocking of pores and difference in atomic weight of nickel and supports. Furthermore, surface area of oxidized catalysts was observed to decrease with increasing nickel loading up to 15 wt% on γ-Al2O3 and levelled off thereafter. The pore volume was also decreased slightly with increasing nickel loading. MD and SM determined from H2 chemisorption results was increased with increasing nickel loading till 10 wt% and levelled off thereafter.| Catalyst | Sa | PVa | MDb | SMb | MH2a | MNH3b | Particle sizec, nm | |
|---|---|---|---|---|---|---|---|---|
| Oxidized | Reduced | |||||||
| a S = surface area, m2 g−1; PV = pore volume, mL g−1; MD = metal dispersion, %; SM = metallic surface area, m2 g−1; MH2 = total amount of H2 consumed mmol g−1 of cat.; MNH3 = total amount of NH3 desorbed, mmol g−1. a Oxidized catalysts. b Reduced catalyst. c Calculated using Debye–Scherer equation. | ||||||||
| Al2O3 | 104 | 8.1 | — | — | 0.35 | — | — | |
| 5NiAl | 88 | 8.3 | 0.21 | 1.42 | 0.54 | 0.23 | — | — |
| 10NiAl | 78 | 8.0 | 0.36 | 2.43 | 2.06 | 0.21 | 9.0 | 9.0 |
| 15NiAl | 68 | 7.9 | 0.15 | 0.98 | 2.91 | 0.17 | 22.0 | 29.0 |
| 20NiAl | 69 | 7.7 | 0.16 | 1.09 | 3.83 | 0.17 | 31.8 | 36.0 |
| SiO2 | 209 | — | — | — | — | — | — | — |
| 15NiSi | 164 | — | 0.29 | 1.96 | 1.93 | — | 19.1 | 26.2 |
| ZSM23 | 318 | — | — | — | — | 1.09 | — | — |
| ZSM55 | 370 | — | — | — | — | 0.64 | — | — |
| 15NiZSM23 | 232 | — | 0.18 | 1.21 | 1.83 | 0.90 | 23.9 | 28.8 |
| 15NiZSM55 | 310 | — | 0.37 | 2.48 | 1.91 | 0.46 | 20.5 | 26.2 |
The XRD patterns of both oxidized and reduced catalysts and pure supports are shown in Fig. 1A and B respectively. Analysis of XRD patterns of oxidized and reduced xNiAl catalysts confirmed presence of bulk nickel oxide (PDF#780643) and nickel (PDF #882326) respectively with nickel loading of 10 wt% or more on γ-Al2O3.26 It was further observed that intensity of bulk nickel oxide (or nickel) peaks was increased with increasing nickel loading on γ-Al2O3. However, only dispersed nickel oxide (or nickel) was present in xNiAl with nickel loading below 10 wt%. XRD patterns of 15NiSi, 15NiZSM23, and 15NiZSM55 also revealed presence of bulk nickel oxide and nickel in oxidized and reduced catalysts respectively. From these results it may be concluded that characteristic bulk nickel peak appeared in XRD pattern of reduced catalysts were most likely originated by reduction of bulk nickel oxide present in the oxidized catalysts. Particle size of the reduced and oxidized catalysts was determined using Debye–Scherer equation. The size of nickel oxide or nickel depends on the nickel loading and nature of support. In case of xNiAl catalysts, the particle size was observed to increase with increasing nickel loading.
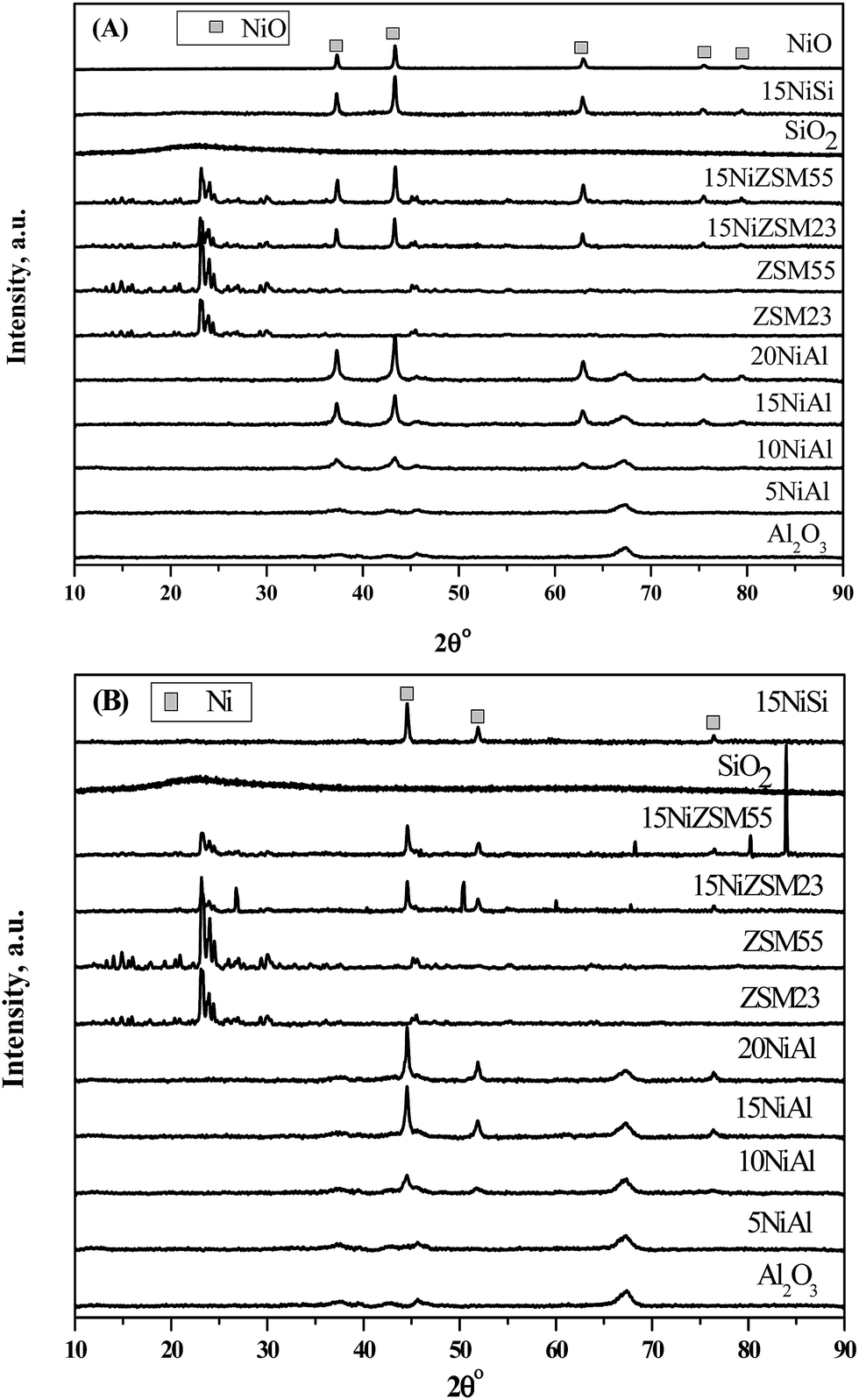 | ||
| Fig. 1 XRD patterns of (A) oxidized and (B) reduced γ-Al2O3, SiO2, ZSM23, and ZSM55 supported nickel catalysts. | ||
The UV-vis DRS spectra of the oxidized catalysts under ambient condition are shown in Fig. 2. The spectra at lower wavelength region are however not shown due to presence of strong charge transfer band that gives very high value of Kubelka Munk function. The band appeared at 280–320 nm (not shown in Fig. 2) for all catalysts is attributed to O2− → Ni+2 charge transfer.27 For xNiAl catalysts, position of this band shifted to higher wavelength with increasing nickel loading. It may be due to increased concentration of bulk nickel oxide. Additional band appeared for xNiAl at 380 and 646 nm are characteristics of nickel aluminate (NiAlO4) which is a solid solution of nickel oxide and γ-Al2O3.27 Evolution of DRS bands at 380, 415 and 715–720 nm was due to bulk nickel oxide with octahedral geometry.27 For xNiAl, these bands were observed for 10 wt% and more nickel loading on γ-Al2O3.
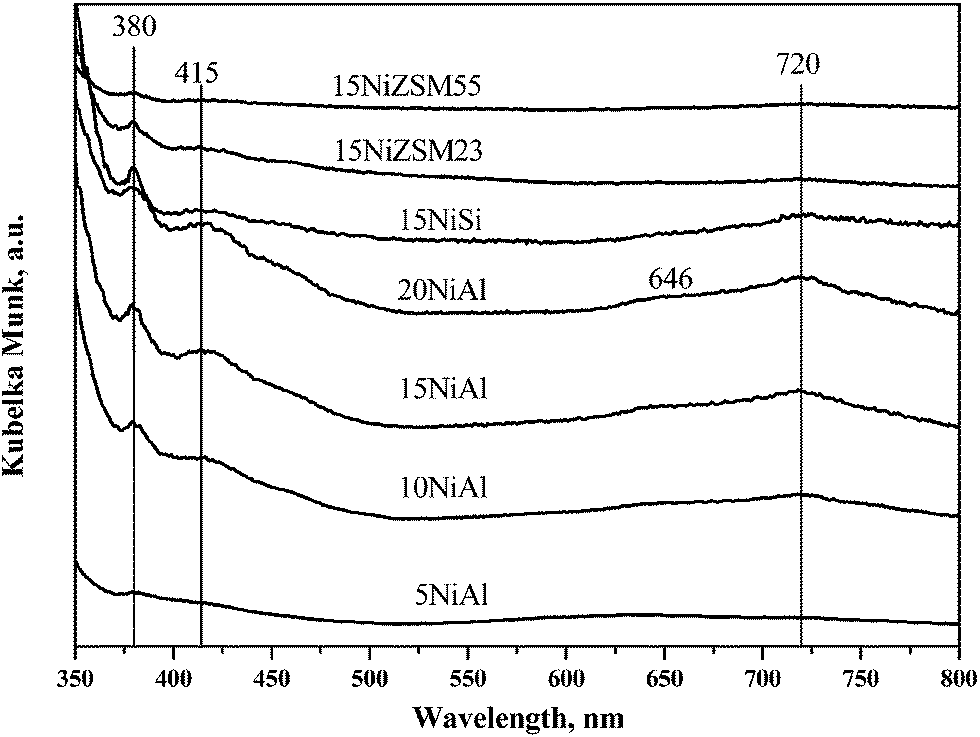 | ||
| Fig. 2 UV-vis spectra of oxidized xNiAl, 15NiSi, 15NiZSM23, and 15NiZSM55 catalysts. | ||
TPR results of the oxidized catalysts are shown in Fig. 3. The peak appeared at 699 K for xNiAl was due to reduction of dispersed nickel oxide possessing strong interaction with the support.26 The peak observed at 627–650 K for 15NiAl and 20NiAl was due to reduction of bulk nickel oxide possessing no or little interaction with support.26 TPR profile of bulk nickel oxide further confirms above argument as shown in the same figure. Moreover, intensity of the reduction peaks corresponding to both dispersed and bulk nickel oxide was increased with increasing nickel loading on γ-Al2O3. The XRD and UV-vis spectroscopy results also confirmed presence of bulk as well as dispersed nickel oxide for 15NiAl and 20NiAl. However, for 10NiAl, the reduction peak corresponding to bulk nickel oxide was appeared as weak shoulder in TPR profiles though it was very prominent in XRD pattern and UV-vis spectra. This might be due to either presence of insignificant amount of bulk nickel oxide in 10NiAl or TPR studies are less sensitive compared to XRD and UV-vis spectroscopy for the detection of specifically small amount of bulk NiO species. The oxidized 15ZSM23 and 15ZSM55 also showed characteristic reduction peaks of both bulk and dispersed nickel oxide. In contrast, 15NiSi showed only a broad reduction peak centered at 605 K. The XRD pattern and UV-vis spectroscopy confirmed presence of bulk nickel oxide in 15NiSi. Thus it can be concluded that reduction of different surface nickel oxide species in 15NiSi might happen within a small range of temperature giving rise to a broad reduction peak centred at 605 K. The H2 consumption during TPR studies was calculated (Table 1) and values were matched closely with theoretical amounts of H2 required to reduce nickel oxide for xNiAl catalysts. Thus, these results indicate that only nickel oxides (dispersed and bulk) are present in the supported nickel catalysts. Contrary to xNiAl, H2 consumption values for 15NiSi, 15NiZSM23, and 15NiZSM55 were lower than theoretically required amounts thereby suggesting the presence of differently coordinated nickel species or a fraction of the nickel may not be accessible to hydrogen during reduction.
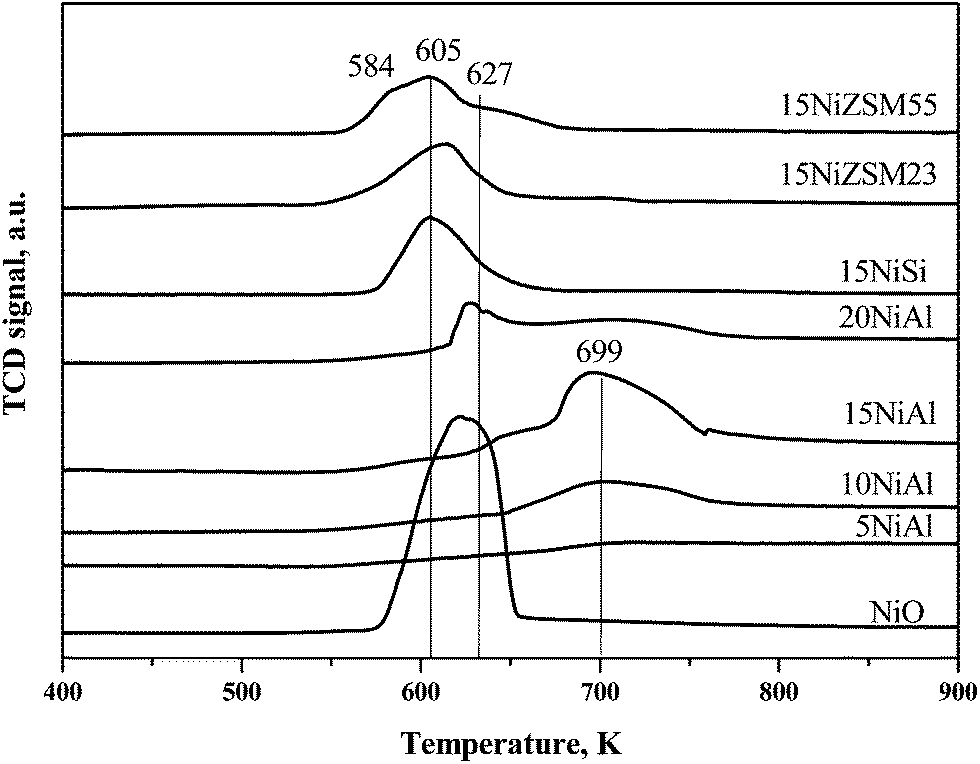 | ||
| Fig. 3 TPR profiles of oxidized xNiAl, 15NiSi, 15NiZSM23, and 15NiZSM55 catalysts. | ||
NH3-TPD results of reduced catalysts and pure supports are shown in Fig. 4 and Table 1. Pure γ-Al2O3 showed a distinct ammonia desorption peak at 445 K corresponding to weak/or moderate acid sites.26 The intensity of this peak was suppressed gradually with increasing nickel loading indicating reduction of acidity with increasing nickel loading on γ-Al2O3.26 The ammonia desorption peak appeared at 913 K and above may be due to dehydroxylation of hydroxyl group present in γ-Al2O3. For zeolites, strength and number of acid sites were significantly more compared to pure γ-Al2O3 with characteristic ammonia desorption peaks at 445–461 K and 645–657 K. The intensity of these peaks was enhanced with decreasing Si/Al ratio suggesting increase of number of acid sites with decreasing Si/Al ratio. Upon deposition of nickel on HZSM-5, intensity of the peaks was decreased significantly together with shifting of peaks to lower temperature (570–578 K). These results indicate that strength and number of acid sites decrease on deposition of nickel on zeolites.
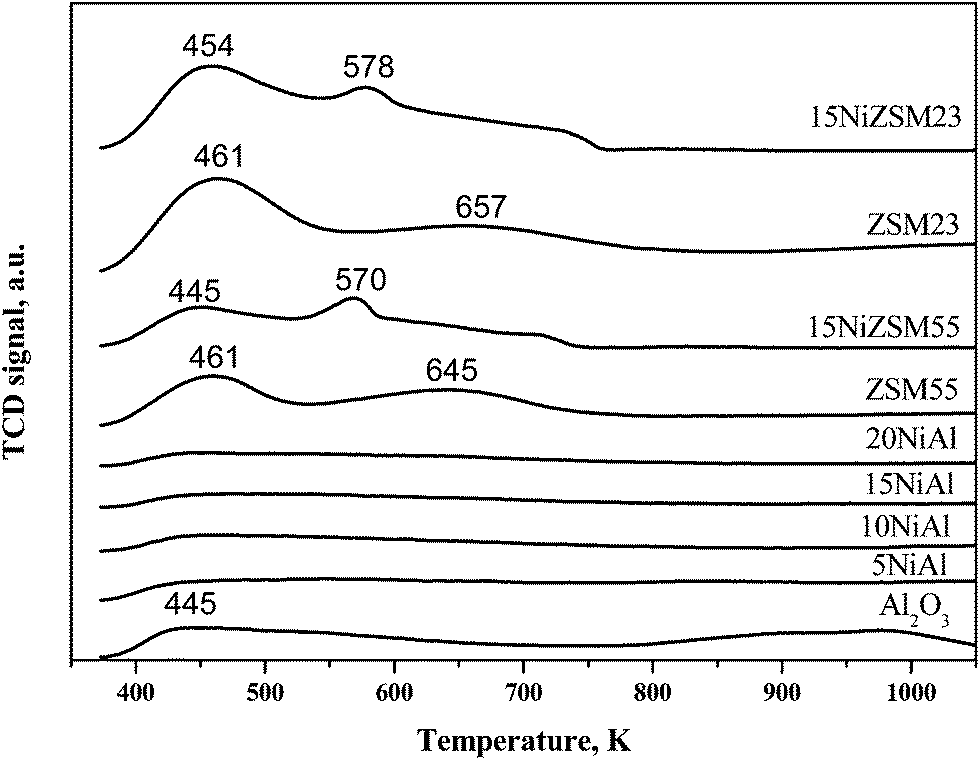 | ||
| Fig. 4 NH3-TPD profiles of xNiAl, 15NiSi, 15NiZSM23, 15NiZSM55 catalysts, and pure supports. | ||
3.2 Reaction mechanism
The n-heptane, n-octane, isomers of heptene (heptenes) and octene (octenes), 1-octanal, di-n-octyl ether (DOE), tetradecane (TETD), and hexadecane (HEXD) were identified by GC-MS in the liquid samples for HDO of 1-octanol (Fig. S1†). Small quantities of octanoic acid (OA) and octyl caprylate (OC, octyl octanoate, C7H15COOC8H17) were also identified in few cases. Furthermore, liquid samples were found to undergo phase separation (aqueous and organic) especially at high conversion of 1-octanol (or freezing the samples below 273 K) demonstrating formation of water as one of the product. A comprehensive reaction mechanism of HDO of 1-octanol was delineated based on products distribution as shown in Scheme 1. 1-Octanol undergoes HDO following two different routes on two different sites of the catalyst: acid and metal.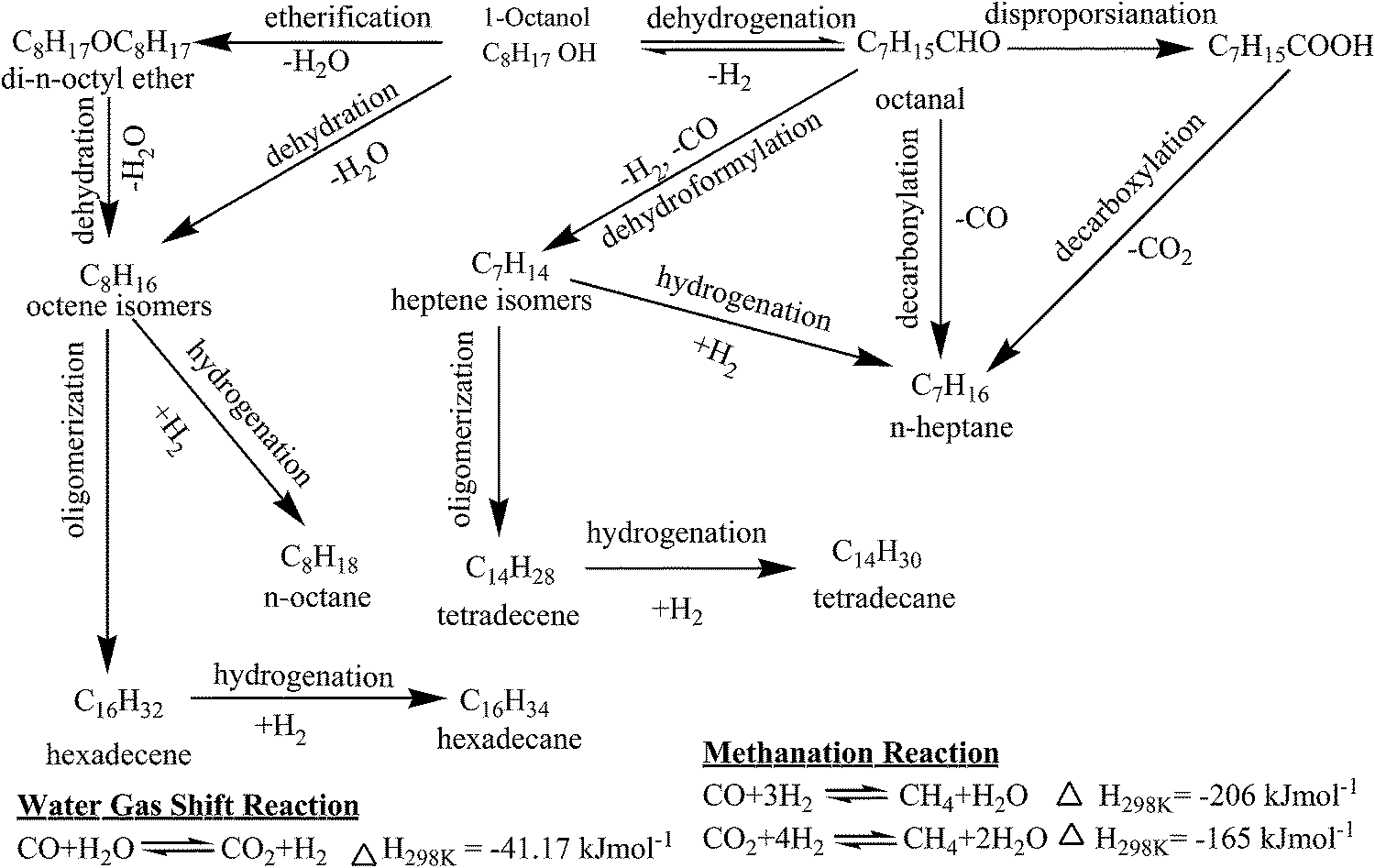 | ||
| Scheme 1 Reaction mechanism for HDO of 1-octanol. | ||
In the first route, 1-octanol undergoes dehydration to octenes either directly or through intermediate etherification reaction via DOE with elimination of one mole of water in each step. The octenes subsequently undergo either hydrogenation to n-octane or oligomerization followed by hydrogenation to HEXD. The acidic sites of catalysts might be responsible for etherification, dehydration, and oligomerization reactions. DOE, n-octane, and octenes were major products for highly acidic pure HZSM-5 and HZSM-5 supported nickel catalysts and their selectivity were also increased with increasing acidity of the catalysts thereby justifying above statements. The dehydration of alcohols, for example ethanol and butanol, was reported previously over acidic catalysts in the temperature range of 498–748 K.28 The oligomerization of olefins, for instance 1-hexene and 1-octene, was also described over various solid acid catalysts such as solid phosphoric acid, ZSM-5, zeolite-Y, and cation exchange resins.29,30 For HDO of 1-heptanol using sulphided NiMo/γ-Al2O3 and CoMo/γ-Al2O3 catalysts, dehydration of alcohol to alkene followed by its hydrogenation to alkane were reported too.21–23
Following second route, 1-octanol undergoes dehydrogenation reaction over active metal sites of the catalysts leading to formation of 1-octanal. The 1-octanal is consequently converted to either n-heptane by decarbonylation reaction or heptenes by dehydroformylation reaction. The heptenes are then either hydrogenated to n-heptane or oligomerized followed by hydrogenated to TETD. The GC analysis of gas sample under nitrogen as carrier gas additionally confirmed formation of both hydrogen and carbon monoxide thereby justifying above claim. Moreover, 1-octanal, n-heptane, and heptenes were primary products for γ-Al2O3 supported nickel catalysts as discussed in subsequent section. Furthermore, their combined selectivity was observed to increase with increasing nickel loading on γ-Al2O3 demonstrating role of metal sites for dehydrogenation, decarbonylation, and dehydroformylation reactions. For catalytic HDO of C3 alcohols, Peng et al. also suggested that alcohol was first dehydrogenated to aldehyde over metal sites.24 The CO underwent water gas shift reaction to give CO2 and H2.
To confirm proposed pathways of conversion of 1-octanal to various products, an independent experiment of HDO of 1-octanal was performed over 15NiAl catalyst using hydrogen as carrier gas (Table S1†). In this particular experiment, n-heptane, 1-octanol, and heptenes were identified as major products together with small amounts of DOE, n-octane, octenes, HEXD, TETD, and OA. The formation of 1-octanol suggests that dehydrogenation of 1-octanol to 1-octanal is a reversible reaction. Small amount of CO2 and CH4 were also detected for HDO of 1-octanol under hydrogen as carrier gas. Formation of CO2 and CH4 may be due to water gas shift reaction and methanation of CO/CO2 respectively as shown in Scheme 1. Formation of OA was however not well understood and might be formed by disproportionation of 1-octanal. The OA formed further undergoes esterification reaction with 1-octanol producing OC. The reaction mechanism is generally confirmed directly through in situ or operando spectroscopic studies. However, it is beyond the scope of present study and left to the researchers as an open challenge problem for future study.
In order to demonstrate the proposed mechanism as generalized one, study was further extended to HDO of 1-heptanol and 1-hexanol over 15NiAl at 518 K. The products distributions for both alcohols were fully in line with proposed mechanism thereby validating the proposed mechanism (Table S1, Fig S2 and S3†). All subsequent studies were performed with 1-octanol only.
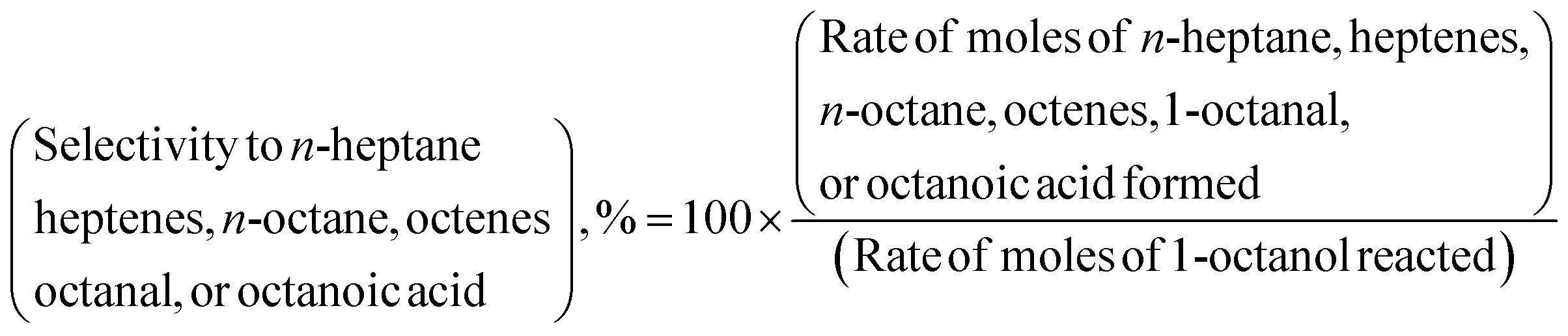 | (1) |
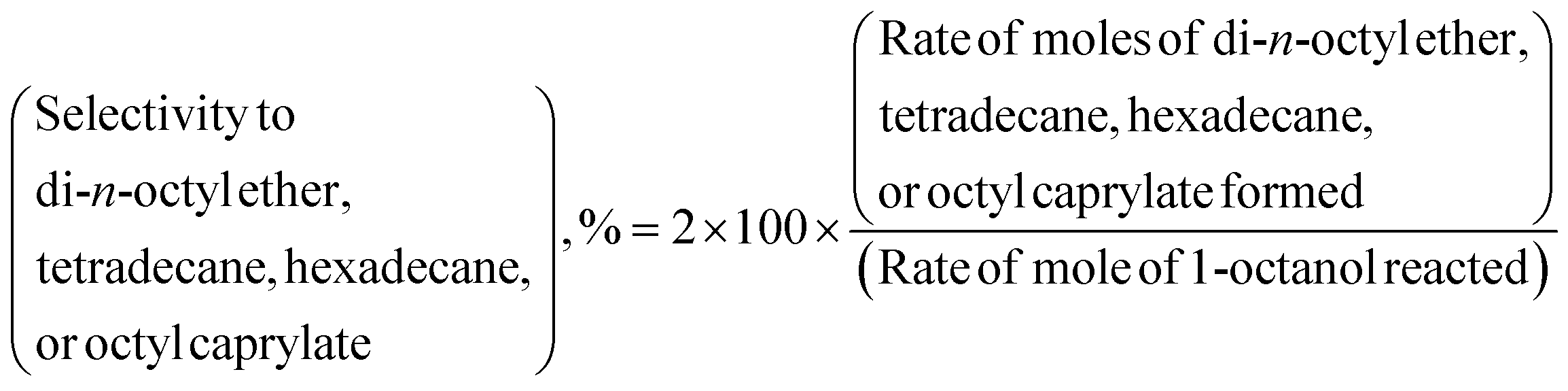 | (2) |
3.3 Reaction studies
The HDO of 1-octanol was investigated in a down-flow fixed-bed reactor over γ-Al2O3, SiO2, ZSM23, and ZSM55 supported nickel catalysts using either nitrogen or hydrogen as carrier gas under wide range of pressures (1–10 bars), nickel loadings on γ-Al2O3 (0–20 wt%), weight hourly space velocities (WHSV) (2.5–9.99 h−1), and temperatures (488–530 K). n-Heptane, heptenes, 1-octanal, DOE, n-octane, and octenes were identified as primary products of HDO of 1-octanol in presence of both nitrogen and hydrogen as carrier gas. The TETD, HEXD, OA, and OC were obtained as minor products of the reaction. The selectivity to n-heptane, heptenes, n-octane, octenes, 1-octanal, and OA used throughout the article is defined by eqn (1). The selectivity to DOE, TETD, HEXD, and OC is defined by eqn (2). It may be noted that all isomers of heptene (1-heptene, 2-heptene, and 3-heptene) and octene (1-octene, 2-octene, and 3-octene) are included within the selectivity to heptenes and octenes respectively.In order to determine critical flow rate of hydrogen, two independent experiments were conducted for two different hydrogen flow rates (20 and 40 mL min−1) maintaining 1-octanol flow rates and WHSV constant. However, 1-octanol conversion and selectivity to products remained practically unaffected with increasing hydrogen flow rate (Table S3†). Therefore, all subsequent experiments were carried out with 20 mL min−1 of hydrogen flow rate.
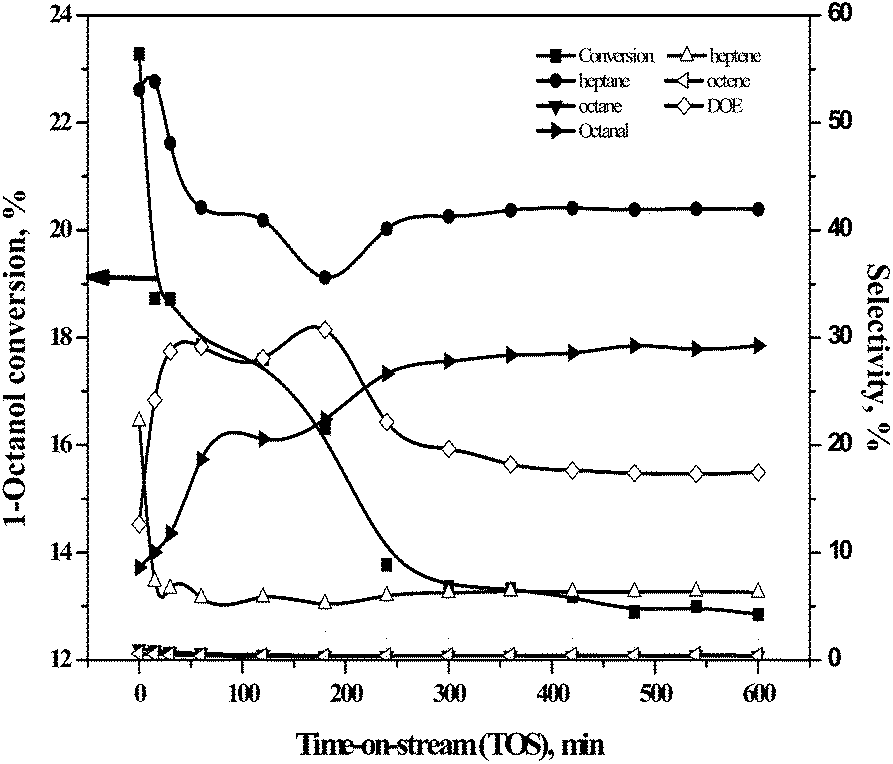 | ||
| Fig. 5 Variation of activity and selectivity of major products for HDO of 1-octanol with TOS. Reaction conditions: catalyst = 15NiAl, temperature = 488 K, pressure = 1 bar, WHSV = 3.99 h−1. | ||
The HDO of 1-octanol was accordingly designed at three different pressures, 1, 5, and 10 bars as presented in Table 2. The conversion of 1-octanol was decreased with increasing pressure indicating that reaction was unfavourable at higher pressure. With increasing pressure, selectivity to 1-octanal, n-heptane, and heptenes were decreased; while selectivity to DOE, n-octane, octenes, TETD, and HEXD were increased. According to Le Chatelier's principle, vapor phase reactions with positive change in number of moles such as dehydrogenation, dehydroformylation, decarbonylation are unfavourable at high pressures and vice versa. This leads to lower selectivity to 1-octanal, n-heptane, and heptenes at higher pressure. The etherification and oligomerization reactions are however favourable at high pressures leading to increasing trends of selectivity to n-octane, octenes, DOE, TETD, and HEXD with pressures. All subsequent experiments were carried out under vapor phase at atmospheric pressure.
| Pressure, bar | Conversion of 1-octanol, % | Selectivity, % | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| n-Heptane | Heptenes | n-Octane | Octenes | 1-Octanal | DOE | HEXD | TETD | OC | OA | ||
| a Reaction conditions: temperature = 503 K, catalyst = 15NiAl, WHSV = 3.99 h−1, TOS = 240 min. | |||||||||||
| 1 | 30.4 | 43.4 | 22.9 | 0.5 | 0.4 | 15.4 | 13.9 | 0.4 | 0.7 | 1.7 | 0.7 |
| 5 | 17.5 | 24.8 | 1.7 | 1.2 | 0.3 | 6.7 | 61.1 | 0.2 | 0.7 | 2.4 | 0.9 |
| 10 | 7.5 | 11.2 | 0.4 | 2.6 | 0.6 | 5.4 | 65.6 | 2.6 | 5.1 | 4.2 | 2.3 |
| Temp., K | Conversion of 1-octanol, % | Selectivity, % | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| n-Heptane | Heptenes | n-Octane | Octenes | 1-Octanal | DOE | HEXD | TETD | OC | OA | ||
| a Reaction conditions: pressure = 1 bar, catalyst = 15NiAl, WHSV = 3.99 h−1, TOS = 240 min. | |||||||||||
| 488 | 13.8 | 40.2 | 6.0 | 0.4 | 0.3 | 26.6 | 22.1 | 0.3 | 1.1 | 2.0 | 1.0 |
| 503 | 30.4 | 43.4 | 22.9 | 0.5 | 0.4 | 15.4 | 13.9 | 0.4 | 0.7 | 1.7 | 0.7 |
| 518 | 45.1 | 50.9 | 24.9 | 0.7 | 0.4 | 10.7 | 10.1 | 0.4 | 0.3 | 1.2 | 0.4 |
| 533 | 53.2 | 57.0 | 23.1 | 1.0 | 0.4 | 9.1 | 7.4 | 0.4 | 0.3 | 1.0 | 0.3 |
| Ni loading, % | Conversion of 1-octanol, % | Selectivity, % | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| n-Heptane | Heptenes | n-Octane | Octenes | 1-Octanal | DOE | HEXD | TETD | OC | OA | ||
| a Reaction conditions: temperature = 503 K, pressure = 1 bar, WHSV = 3.99 h−1, TOS = 240 min. | |||||||||||
| 0 | 9.8 | 0 | 0 | 0 | 17.5 | 0.8 | 81.7 | 0 | 0 | 0 | 0 |
| 5 | 10.4 | 12.1 | 15.5 | 1.7 | 2.4 | 28.1 | 32.4 | 0.5 | 1.5 | 4.3 | 1.5 |
| 10 | 16.7 | 18.6 | 20.3 | 1.1 | 0.9 | 28.4 | 25.5 | 0.4 | 1.2 | 2.4 | 1.2 |
| 15 | 30.4 | 43.4 | 22.9 | 0.5 | 0.4 | 15.4 | 13.9 | 0.4 | 0.7 | 1.7 | 0.7 |
| 20 | 20.7 | 47.7 | 14.4 | 1.3 | 0.3 | 18.2 | 15.5 | 0.3 | 0.7 | 0.9 | 0.7 |
In order to delineate role of supports on the reaction, HDO of 1-octanol was conducted over pure supports (γ-Al2O3, SiO2, ZSM23 and ZSM55) as well (Tables 4 and 5). 1-Octanol mainly undergoes dehydration and etherification reaction in presence of pure supports resulting in formation of n-octane, octenes, and DOE as foremost products together with only small quantity of 1-octanal. The relative proportion of dehydration and etherification products strongly depends on strength and nature of acid sites (Lewis and Bronstead). Strongly acidic supports promote dehydration of alcohols resulting in the formation of corresponding olefins as major products. In general, Lewis acid sites of γ-Al2O3 are responsible for etherification reaction of 1-octanol yielding DOE as major product together with lesser amount of octenes. The octenes thus formed however cannot easily undergo hydrogenation reaction even in presence of hydrogen due to non-availability of active metal sites in the catalyst. Hence, poor selectivity to alkanes was observed for pure supports.
| Catalysts | Conversion of 1-octanol, % | Selectivity, % | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| n-Heptane | Heptenes | n-Octane | Octenes | 1-Octanal | DOE | HEXD | TETD | OC | OA | ||
| a Reaction conditions: temperature = 518 K, pressure = 1 bar, WHSV = 3.99 h−1, TOS = 240 min. | |||||||||||
| SiO2 | 1.7 | 9.3 | 0 | 12.4 | 54.9 | 9.7 | 2.9 | 0 | 0 | 0.2 | 10.6 |
| ZSM23 | 55.6 | 4.0 | 0 | 16.4 | 77.8 | 1.0 | 0.8 | 0 | 0 | 0 | 0 |
| ZSM55 | 87.4 | 2.3 | 0 | 9.8 | 85.5 | 1.0 | 1.5 | 0 | 0 | 0 | 0 |
| 15NiAl | 45.1 | 50.9 | 24.9 | 0.7 | 0.4 | 10.7 | 10.1 | 0.4 | 0.3 | 1.2 | 0.4 |
| 15NiSi | 43 | 52.7 | 32.8 | 0.3 | 0.2 | 10.8 | 2.6 | 0 | 0 | 0 | 0.6 |
| 15NiZSM23 | 40.2 | 8.2 | 0 | 5.9 | 73.2 | 6.2 | 6.5 | 0 | 0 | 0 | 0 |
| 15NiZSM55 | 61.5 | 10.6 | 0 | 4.0 | 69.5 | 6.2 | 6.9 | 2.6 | 0 | 0.1 | 0.1 |
The 15 wt% nickel loaded on γ-Al2O3, SiO2, ZSM23, and ZSM55 catalysts were also tested for HDO of 1-octanol (Table 5). As observed from the table, C8 hydrocarbons and DOE were primary products over highly acidic catalysts (15ZSM23 and 15ZSM55).31 On the other hand, C7 hydrocarbons and DOE were main products for 15NiAl and 15NiSi. From these results it may be concluded that HDO of 1-octanol mainly proceeds through dehydration pathway in presence of acidic catalysts and dehydrogenation pathway for metallic catalysts.
| WHSV, h−1 | Conversion of 1-octanol, % | Selectivity, % | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| n-Heptane | Heptenes | n-Octane | Octenes | 1-Octanal | DOE | HEXD | TETD | OC | OA | ||
| a Reaction conditions: temperature = 503 K, pressure = 1 bar, catalyst = 15NiAl, TOS = 240 min. | |||||||||||
| 10.0 | 15.2 | 49.4 | 21.4 | 0.7 | 0.4 | 14.8 | 9.5 | 0.4 | 1.1 | 1.1 | 1.2 |
| 5.0 | 25.0 | 45.0 | 22.3 | 0.6 | 0.4 | 16.5 | 11.7 | 0.5 | 0.6 | 1.8 | 0.6 |
| 3.3 | 31.7 | 45.2 | 23.4 | 0.6 | 0.6 | 15.1 | 12.2 | 0.4 | 0.6 | 1.3 | 0.6 |
| 2.5 | 37.2 | 41.9 | 21.8 | 0.6 | 0.4 | 12.5 | 18.5 | 0.6 | 0.6 | 2.6 | 0.5 |
4 Conclusions
The HDO of 1-octanol was investigated in a down-flow fixed-bed reactor using γ-Al2O3, SiO2, and HZSM-5 supported nickel catalysts prepared by incipient wetness impregnation method. The prepared catalysts together with pure supports were characterized by BET, XRD, UV-vis, TPR, TPD, and H2 chemisorption. The supported nickel (or nickel oxide) catalysts were observed to be associated with bulk nickel (or nickel oxide) as well beyond 10 wt% nickel loading on the supports. The strength and number of acid sites of the supported nickel catalysts were somewhat lesser compared to their respective pure supports. HDO of 1-octanol follows two different pathways depending on nature of the catalysts. HDO reaction proceeds largely through dehydration pathway over acidic catalysts with formation of DOE, isomers of octene, and n-octane as primary products. The dehydrogenation route becomes dominant over metallic catalysts leading to formation of 1-octanal, isomers of heptene, and n-heptane as predominating products. The higher conversion of 1-octanol and selectivity to alkanes was observed for hydrogen as a carrier gas compared to that of nitrogen. The conversion of 1-octanol was increased with increasing nickel loading on γ-Al2O3 and temperatures and decreasing pressure and WHSV. The combined selectivity to n-heptane, heptenes, and 1-octanal was increased with increasing nickel loading on γ-Al2O3 and temperatures and decreasing pressure. The combined selectivity to n-octane, octenes, and DOE was however observed to follow reverse trend.Nomenclatures
| HDO | Hydrodeoxygenation |
| WHSV | Weight hourly space velocity |
| xNiAl | x wt% nickel supported on γ-Al2O3 |
| 15NiSi | 15 wt% nickel supported on SiO2 |
| ZSMy | HZSM-5 with Si/Al = y, where y = 23 and 55 |
| 15NiZSMy | 15 wt% nickel supported on HZSM-5 |
| TETD | Tetradecane |
| HEXD | Hexadecane |
| DOE | Di-n-octyl ether |
| OC | Octyl caprylate |
| OA | Octanoic acid |
| TOS | Time-on-stream |
Acknowledgements
The authors gratefully acknowledge the financial support from the Department of Science and Technology (DST), India.References
- Introduction to Chemicals from Biomass, ed. J. H. Clark and F. E. I. Deswarte, John Wiley and Sons, Ltd, 2008 Search PubMed.
- H. S. Choi, Y. S. Choi and H. C. Park, Renewable Energy, 2012, 42, 131–135 CrossRef CAS PubMed.
- D. Mohan, C. U. Pittman and P. H. Steele, Energy Fuels, 2006, 20, 848–889 CrossRef CAS.
- S. Fernando, S. Adhikari, C. Chandrapal and N. Murali, Energy Fuels, 2006, 20, 1727–1737 CrossRef CAS.
- G. W. Huber, S. Iborra and A. Corma, Chem Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- A. V. Bridgwater, Chem. Eng. J., 2003, 91, 87–102 CrossRef CAS.
- S. Czernik and A. V. Bridgwater, Energy Fuels, 2004, 12, 590–598 CrossRef.
- D. Hong, S. J. Miller, K. Agrawal and C. W. Jones, Chem. Commun., 2010, 46, 1038–1040 RSC.
- E. Furimsky, Appl. Catal., A, 2000, 199, 147–190 CrossRef CAS.
- Q. Bua, H. Leia, A. H. Zacherb, L. Wanga, S. Rena, J. Lianga, Y. Weia, Y. Liua, J. Tanga, Q. Zhanga and R. Ruan, Bioresour. Technol., 2012, 124, 470–477 CrossRef PubMed.
- H. Wang, J. Male and Y. Wang, ACS Catal., 2013, 3, 1047–1070 CrossRef CAS.
- M. Saidi, F. Samimi, D. Karimipourfard, T. Nimmanwudipong, B. C. Gates and M. R. Rahimpour, Energy Environ. Sci., 2014, 7, 103–129 CAS.
- Y. Wang, J. Wu and S. Wang, RSC Adv., 2013, 3, 12635–12640 RSC.
- B. Donnis, R. G. Egeberg, P. Blom and K. G. Knudsen, Top. Catal., 2009, 52, 229–240 CrossRef CAS.
- D. Procházková, P. Zámostný, M. Bejblová, L. Červený and J. Čejka, Appl. Catal. A: Gen., 2007, 332, 56–64 CrossRef PubMed.
- M. R. D. Brimont, C. Dupont, A. Daudin, C. Geantet and P. Raybaud, J. Catal., 2012, 286, 153–164 CrossRef PubMed.
- S. Sitthisa, T. Sooknoi, Y. Ma, P. B. Balbuena and D. E. Resasco, J. Catal., 2011, 277, 1–13 CrossRef CAS PubMed.
- S. Sitthisa and D. E. Resasco, Catal. Lett., 2011, 141, 784–791 CrossRef CAS.
- J. Huang, W. Long, P. K. Agrawal and C. W. Jones, J. Phys. Chem. C, 2009, 113, 16702–16710 CAS.
- M. A. Alotaibi, E. F. Kozhevnikova and I. V. Kozhevnikov, Chem. Commun., 2012, 48, 7194–7196 RSC.
- E.-M. Ryymin, M. L. Honkela, T.-R. Viljava and A. O. I. Krause, Appl. Catal., A, 2009, 358, 42–48 CrossRef CAS PubMed.
- O. Şenol, E.-M. Ryymin, T.-R. Viljava and A. O. I. Krause, J. Mol. Catal. A: Chem., 2007, 268, 1–8 CrossRef PubMed.
- O. Şenol, T.-R. Viljava and A. O. I. Krause, Appl. Catal., A, 2007, 326, 236–244 CrossRef PubMed.
- B. Peng, C. Zhao, I. Mejía-Centeno, G. A. Fuentes, A. Jentys and J. A. Lercher, Catal. Today, 2012, 183, 3–9 CrossRef CAS PubMed.
- V. Dhanala, S. K. Maity and D. Shee, RSC Adv., 2013, 3, 24521–24529 RSC.
- G. Sadanandam, K. Ramya, D. B. Kishore, V. Durgakumari, M. Subrahmanyam and K. V. R. Chary, RSC Adv., 2014, 4, 32429–32437 RSC.
- B. Scheffer, J. J. Heijeinga and J. A. Moulijn, J. Phys. Chem., 1987, 91, 4752–4759 CrossRef CAS.
- X. Zhang, R. Wang, X. Yang and F. Zhang, Microporous Mesoporous Mater., 2008, 116, 210–215 CrossRef CAS PubMed.
- A. Klerk, Ind. Eng. Chem. Res., 2005, 44, 3887–3893 CrossRef.
- R. Bringué, M. Cadenas, C. Fité, M. Iborra and F. Cunill, Chem. Eng. J., 2012, 207–208, 226–234 CrossRef PubMed.
- P. Kumar, S. R. Yenumala, S. K. Maity and D. Shee, Appl. Catal., A, 2014, 471, 28–38 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: HDO of 1-octanal, 1-octanol, 1-heptanol and 1-hexanol; Effects of H2 flow rate on HDO of 1-octanol; vapour pressure of pure components; GCMS chromatogram of liquid sample HDO of 1-octanol, 1-hexanol and 1-heptanol. See DOI: 10.1039/c4ra06826b |
| This journal is © The Royal Society of Chemistry 2014 |