
Computational studies on interparticle forces between nanoellipsoids†
Weifu Sun‡
*a,
Qinghua Zengb and
Aibing Yu*ac
aLaboratory for Simulation and Modeling of Particulate Systems, School of Materials Science and Engineering, The University of New South Wales, Sydney, NSW 2052, Australia. E-mail: weifu.sun518@gmail.com; weifu.sun@sydney.edu.au
bSchool of Computing, Engineering and Mathematics, University of Western Sydney, Penrith, NSW 2751, Australia
cDepartment of Chemical Engineering, Monash University, Clayton, Victoria 3800, Australia. E-mail: aibing.yu@monash.edu; Fax: +61-3-9905-5686; Tel: +61-3-9905-0582
First published on 13th August 2014
Abstract
The adapted continuum models pertinent to ellipsoidal microparticles do not generally hold at the nanoscale due to the approximations apart from the surface effects and the neglect of atomic discrete structure. The governing equation of describing the interactions, non-contact forces in particular, between ellipsoidal nanoparticles is lacking. In this work, the interaction forces including van der Waals (vdW) attraction, Born repulsion and mechanical contact forces between nanoellipsoids are studied by molecular dynamics (MD) simulation and compared with those predicted by the adapted continuum models (Hamaker or Hertz model). The results show that the interaction forces between ellipsoidal nanoparticles are complicated and the ratios of interaction forces obtained from the MD simulations to those from the adapted Hamaker equations are dependent on surface separation, particle size, aspect ratio and configurations. Under different configurations, two formulas have been proposed for vdW attraction and Born repulsion forces. In particular, under parallel configuration, both the vdW attraction and Born repulsion forces between nanoellipsoids show an obvious periodic variation stemming from the step-like atomic structure and can be described by a second-order Fourier expansion and correspondingly another two relatively more accurate formulas are proposed for vdW attraction and Born repulsion forces. Moreover, the mechanical contact force between ellipsoidal nanoparticles at low compression still can be described by the Hertz model. This work can provide quantitative insights into interaction forces between nano-ellipsoids and should be useful in the applications where ellipsoidal particles are involved, such as self-assembly by virtue of inter-particle or external forces.
1. Introduction
In practice, most particles are not perfect spheres as usually treated in many theoretical and experimental studies. Ellipsoidal particles are one of the most common non-spherical particles, which are of general interest for practical applications.1–4 In particular, the ellipsoids, as anisotropic particles, provide a convenient approach by shapes to produce interparticle capillary interaction to suppress the coffee-ring effect4 in a bid to achieve uniform deposition of particles or coatings in many applications including printing,5 biology6 and complex assembly.7,8 Nanoparticles in the range of 1–10 nm bear unique electronic, optical, and mechanical properties different from those of their bulk materials because of the significant contribution of surface atoms, such as quantum dots.9 The assembly10 of nanoparticles is a key step for their application as nanodevices. The properties of self-assembly structures strongly depend on the interaction forces,11,12 particle shape,13 nanoparticle ordering processes and, of course, the properties of the nanoparticle building blocks themselves.14 Thus, to have a full understanding of the various inter-ellipsoid forces (e.g., van der Waals (vdW) attraction, Born repulsion and contact forces) is important to achieve a better control over their ordering into larger scale nanostructured materials. Such forces also play an important role in understanding natural phenomena15,16 and industrial processes.17However, the continuum models such as Hamaker approach18 and Hertz continuum model19 do not generally apply at the nanoscale due to the neglect of atomic discrete structure,20,21 surface effects, the underestimated number density of atoms or the neglected intermolecular forces. On one hand, the continuum Hamaker approach treats the particles as rigid (incompressible) bodies without considering the atoms' vibration, bond's torsion, inversion, etc., implying that the particles will not deform and this is contrary to the reality; on the other hand, in the Hamaker approach, particles are assumed or treated as a continuum medium with uniform (density) distribution of atoms. But in essence, the structure of nanoparticle is not only ‘soft’, but also discrete. For large ellipsoidal particles, the interaction potentials between them are usually evaluated by Hamaker approach18,22 or Gay–Berne (GB) approach.23 GB approach can be applied to anisotropic interactions in a computationally efficient way but lacks a clear microscopic interpretation.24 In contrast, Hamaker approach can be adapted to estimate the interaction potentials between micro/macro-ellipsoids of arbitrary size and shape with a well-defined microscopic interpretation.25 However, the adapted Hamaker approach could not be directly applied to nano-ellipsoids owing to the approximations apart from the other same demerits as nanospheres, brought by the Hamaker approach, i.e., the neglect of the nature of discrete atomic structures and surface effects.20,21,26,27 The governing equations of describing interaction forces between nanoellipsoids remain unknown, which in turn can adversely affect our capability of understanding natural phenomena and material design of nanodevices. Thus a better prediction of interaction forces between ellipsoidal nanoparticles calls for detailed investigation. One should bear in mind that even at the macroscale, different from spherical particles, the equations used to describe interaction forces between ellipsoidal particles become much more complicated than spheres, depending on particle size, aspect ratio and orientation, etc.
For macro/micro-sized particles, the interaction forces between ultrafine cohesive frictional particles can be studied by experimental techniques, such as atomic force microscopy (AFM).28–30 But for nanoparticles below 10 nm, at this stage of development, the accuracy of AFM measurement, especially in the lateral direction, is critically dependent on factors such as sensitive spring constant, precision calibration of the cantilever, etc. Moreover, its accuracy is also affected by many other factors such as sample contamination, water meniscus formation, laser interference and the coupling between the vertical and lateral deflections, etc. How to accurately quantify interaction forces at the ultra-small nanoscale is still a challenge in the research community. Thus molecular dynamics (MD) simulation, as an alternative tool to experimental techniques, has a valuable role to play in providing almost exact results.31–37
Silicon, both as a substrate for compatibility with semiconductor processing equipment and as a structural material,38 is the most common single material used in the electronics industry and has applications in areas such as NEMS devices,39 photonic and biotechnology.40 In present work, MD simulations will be applied to study the interaction forces between ellipsoidal nanoparticles based on silicon materials, including the vdW attraction, Born repulsion and mechanical contact forces. This report is organized as follows. The simulation method and conditions will be introduced in Section 2. The effects of particle size, aspect ratio and configuration on interaction forces between nanoellipsoids will be examined in Section 3, in which a comparison will be made to the Hamaker and Hertz predictions, and formulas will be developed. The main conclusions are summarized in the last section.
2. Simulation conditions and method
MD simulations are performed by Materials Studio package using COMPASS force field,41 which consists of a Lennard-Jones (LJ) potential function for Born repulsion and vdW attraction between non-bonded atoms as well as valence interactions such as bond stretch, angle bending, angle torsion, angle inversion, etc., between bonded atoms. The cutoff distance employed in the present MD simulations is set to be as large as 100 nm to completely avoid the errors possibly introduced by the cutoff distance. The Hamaker constant for silicon is calculated based on the parameters used in COMPASS. The values of σi and εi for silicon are 4.45 Å and 0.198 kcal mol−1, respectively.41,42 Thus the Hamaker constant A can be estimated to be about 1.48 × 10−19 J (A = π2C/v2, where C = 3εSi–SiσSi–Si6 is the vdW attraction interaction parameter, v = 4π(σSi–Si/2)3/3 is the atom volume of about 46.140 Å3) according to the method inherent in the Hamaker approach.While the simulation method has been detailed elsewhere,32,33 the major simulation procedures are outlined below:
(i) Silicon nanoellipsoids with different principal radii (a, b, c) and a condition of a = c < b, are first carved out of their cubic silicon bulk counterparts by specifying the three different principal radii, and each individual nanoellispoid is fully relaxed using the NVT ensemble (i.e., constant number of atoms, constant volume and constant temperature) at 300 K. Note that, different from silicon carbide nanospheres in the previous work, which is carved out of their triclinic bulk counterpart,43 the silicon nanoellipsoids are carved out of their diamond-like cubic structure.
(ii) Then, two identical nanoellipsoids are placed at a certain surface separation distance apart, which is normally at least twice greater than particle's principal radius along the particular direction to ensure that the fully relaxed initial configuration is not affected by the interactions between them, followed by geometry optimization and MD simulations using the NVE ensemble (i.e., constant number of atoms, constant volume and constant energy) and the velocity Verlet integration algorithm with a timestep of 1 fs (Δt = 1.0 × 10−15 s). The two fully relaxed nanoellipsoids are allowed to move toward each other at an equal but opposing initial velocity. Besides, it is worth mentioning that the possible surface diffusion of atoms on the surface of particle is not observed even when the two particles are closely put together (Fig. S-1, ESI†).
(iii) Energies, forces, coordinates and other information are recorded every 100 steps in an output trajectory file and then correlated to the surface separation distance d between the nanoellipsoids.
The silicon nanoellipsoids of (1.25, 2.50, 1.25), (2.50, 5.00, 2.50), (3.75, 7.50, 3.75), (1.25, 5.00, 1.25) and (3.75, 5.00, 3.75) in radius (unit: nm) contains 833, 6498, 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 067, 1650 and 14682 silicon atoms, respectively. After running the MD simulations using the NVE ensemble, the properties of atoms such as coordinates, forces and velocities along each direction (i.e., X-, Y- or Z-axis) can be separately obtained from the trajectory files. Of note, in our case, in order to ensure normal impact (that is central collision) meanwhile to minimize the dynamic effect,44 4.0 Å ps−1 (i.e., 400 m s−1) along the X-axis of each atom in the nanoellipsoid was specified while 0 Å ps−1 was specified to the other two directions. The use of high impact speed may have its pros and cons. On one hand, since the interparticle potentials such as vdW attractive and Born repulsive potentials are as a function of relative positions of atoms contained in each particle, thus strictly speaking, different impact velocity can have an influence on the interparticle potentials at close separation since the surface atoms do not have enough time to adjust positions to reach a potential minimum. However, in the previous work based on silica nanospheres, the results show that the inter-particle potentials are almost independent of impact velocity.33 This may indicate that the effect of the slight change of atom's positions at close separation on interaction potentials is limited. On the other hand, the use of high speed can ensure the head-collision and decrease the effect of non-central collision to a minimum. Another benefit is that while speeding up the dynamic process, at high impact velocity certain deformation of particle can be created whereas at low impact velocity no obvious deformation may be observed.
067, 1650 and 14682 silicon atoms, respectively. After running the MD simulations using the NVE ensemble, the properties of atoms such as coordinates, forces and velocities along each direction (i.e., X-, Y- or Z-axis) can be separately obtained from the trajectory files. Of note, in our case, in order to ensure normal impact (that is central collision) meanwhile to minimize the dynamic effect,44 4.0 Å ps−1 (i.e., 400 m s−1) along the X-axis of each atom in the nanoellipsoid was specified while 0 Å ps−1 was specified to the other two directions. The use of high impact speed may have its pros and cons. On one hand, since the interparticle potentials such as vdW attractive and Born repulsive potentials are as a function of relative positions of atoms contained in each particle, thus strictly speaking, different impact velocity can have an influence on the interparticle potentials at close separation since the surface atoms do not have enough time to adjust positions to reach a potential minimum. However, in the previous work based on silica nanospheres, the results show that the inter-particle potentials are almost independent of impact velocity.33 This may indicate that the effect of the slight change of atom's positions at close separation on interaction potentials is limited. On the other hand, the use of high speed can ensure the head-collision and decrease the effect of non-central collision to a minimum. Another benefit is that while speeding up the dynamic process, at high impact velocity certain deformation of particle can be created whereas at low impact velocity no obvious deformation may be observed.
A typical sequence of a pair of colliding ellipsoidal nanoparticles is shown in Fig. 1. They move toward each other to a position (Fig. 1a) at an initial relative velocity of Vr,0 = 400 m s−1, and reach in close proximity (Fig. 1c and d). Then, overlapping occurs (Fig. 1e), followed by the departure process (Fig. 1f–h). A positive relative velocity (Vr > 0) denotes the approach toward each other while a negative value (Vr < 0) represents the departure from each other. The surface separation (d = r − R1 − R2) denotes the corresponding shortest surface-to-surface separation, where r is the centre-to-centre separation distance, R1 and R2 are the defined principal radii of nanoellipsoids 1 and 2, respectively, along the head-on direction.
 | ||
| Fig. 1 Sequential snapshots of a typical head-on collision and departure of two silicon nanoellipsoids of (2.5 nm, 5.0 nm, 2.5 nm). (a): 32.6 ps, (b) 33.8 ps, (c): 36.3 ps, (d): 37.1 ps, (e): 37.6 ps, (f): 39.1 ps, (g): 40.5 ps and (h): 42.3 ps. The initial relative velocity is Vr,0 = 400 m s−1. | ||
During the head-on collision, the relative velocity Vr between the two interacting nanoellipsoids as a function of the time is displayed in Fig. 2. It can be observed that during the approach process, the relative velocity Vr gradually increases under the influence of the interparticle vdW attractive forces and reaches a peak at around 37 ps (i.e., at close separation corresponding to Fig. 1d). Then it decreases sharply to almost zero because of the strong short-ranged Born repulsion forces after contact deformation. Finally during the rebounding and departure process, the relative velocity between them first increases sharply under the influence of Born repulsion force to a peak at about 41 ps, following by a slight reduction and level-off. However, in general, the relative velocity at the similar surface separation apart in the departure process becomes smaller than those in the approach process due to the energy loss.43
 | ||
| Fig. 2 The relative velocity Vr between two parallel silicon nanoellipsoids of (2.5, 5.0, 2.5) as a function of the time. The initial relative velocity Vr,0 is 400 m s−1 and the initial surface separation is 15.0 nm along Y-axis. | ||
The interaction potential energies and their individual potential contributions (e.g., vdW attraction, Born repulsion) can be calculated from the potential energies of total and individual particles.32,33 Then, by differentiating the interaction potentials with respect to their surface separation, the corresponding interaction forces between nanoellipsoids can be obtained.
It is worth mentioning that since the process simulated here, i.e., head-on collision, is a non-equilibrium dynamic process, there is always a change in temperature when two particles get close no matter which ensemble (NVE or NVT) is used. But it is demonstrated that the temperature and ensemble has little effect on the MD simulated results, which can be ignored.32,33 This also implies that small disturbance in positions of atoms, for example, stemming from thermal vibrations within nanoparticles, does not affect the simulated results. Another concern is perhaps about the many-body effect among atoms, which has been at least partly considered in formulating the inter-nanoparticle forces since the possible interactions among atoms in the nanospheres have been accounted through the COMPASS force field (e.g., valence terms including bending, torsion, inversion, etc.); on the other hand, it was reported that many-body effects can be ignored for shape-isotropic nanocluster,45 thus MD simulations could provide accurate results of interaction forces. Besides, it is worth mentioning that the COMPASS force field was developed based on some quantum mechanics calculations46–49 (i.e., ab initio calculation herein) and it should provide relatively accurate estimation of materials properties. During the dynamic simulation process, in a bid to make the two nanoellipsoids move towards each other, thus all the atoms within one nanoellipsoid was assigned an initial velocity of 200 m s−1 while the atoms within the other nanoellipsoid was assigned an equal but opposite initial velocity to make them move towards each other at a relative velocity of 400 m s−1.
Note that in this work the vdW attraction, Born repulsion (i.e., the short range repulsion force) and mechanical contact forces will be examined. It is easily understood that the former two types of non-contact forces stem from the LJ potential, whereas the origin of the mechanical contact forces can be ascribed to arise from valence interactions between bonded atoms such as bond stretch, angle bending, angle torsion, angle inversion. Because there is always a force constant to describe these terms in their individual expressions in COMPASS force field.50 These valence interactions should be related to macro-mechanical properties, leading to mechanical contact force pertinent to contact deformation. Therefore, in addition to the interparticle vdW attraction and Born repulsion forces, mechanical contact force Fc will arise upon the contact deformation of two nanoparticles.
3. Results and discussion
To develop the formulas for nanoellipsoids, interaction forces will be studied in terms of particle size, aspect ratio and configuration. In addition, the particle radius and different parameters of contacting nanoellipsoids will be defined and calculated first, such as radius of principal curvature (R′1 and R′′1), and equivalent radius Re. Note that as for nanoellipsoids, different from spherical particles, there are three principal radii along the three different directions and the radial distance from the particle centre is varied and not a constant and it is hard to distinguish “surface” from “core” in terms of structural point as done for spheres, and also quite difficult to obtain one surface roughness through the characterization of ellipsoid. Although ellipsoidal particle has three principal radii, their surface separation and principal radius along one particular direction can still be calculated by the same approach from the corresponding nanospheres.32,33 When calculating the surface separation, to be consistent with the previous work, the defined principal radius along one particular direction can be obtained based on the corresponding nanospheres with radius equal to the principal radius in the particular direction. Therefore, the corresponding Si nanospheres were characterized and the surface roughness obtained from Si nanospheres will be used in this work, e.g., eqn (5), (10) and (13).The calculated results show that the defined particle radii R corresponding to the cut radii R0 of 1.25, 2.50, 3.75 and 5.00 nm are 1.156, 2.368, 3.604 and 4.849 nm, respectively; the rms is about 0.62 ± 0.03 Å, δ is about 1.55 ± 0.14 Å, δMax is about 2.09 ± 0.43 Å. The defined particle radius R will be used in calculating surface separation d. One illustration of the above defined structural parameters are shown in Fig. S-2 and Table S-1, ESI† and the main nomenclature for symbols used in this work is summarized in Table S-2, ESI.†
The equations used to determine the structural parameters of two ellipsoids with their axes inclined to each other by an angle α, are given by51
 | (1) |
 | (2) |
At apex of (a, 0, 0), the principal radii of curvature are calculated by R′1 = b2/a and R′′1 = c2/a; similarly, at apex of (0, b, 0), R′2 = a2/b and R′′2 = c2/b; at apex of (0, 0, c) R′3 = a2/c and R′′3 = b2/c. Note that the ellipsoids in this work are probate spheroids with two equal principal radii and aspect ratio greater than 1.
3.1. Size dependence
MD simulations are performed on a side by side impact of two parallel silicon spheroids. The radii of two principal curvatures at apex (a, 0, 0) are calculated (Table 1) and then using eqn (1) and (2), the parameters A, B, aspect ratio , orientation parameter χ12η12 and the overall equivalent radii Re are calculated (Table 2). Note that the radii of two principal curvatures at apex (a, 0, 0) of the second ellipsoid are equal to those of the first one.
, orientation parameter χ12η12 and the overall equivalent radii Re are calculated (Table 2). Note that the radii of two principal curvatures at apex (a, 0, 0) of the second ellipsoid are equal to those of the first one.
| Apex | (1.25, 0, 0) | (2.5, 0, 0) | (3.75, 0, 0) |
|---|---|---|---|
| R′1 | 5.0 | 10.0 | 15.0 |
| R′′1 | 1.25 | 2.5 | 3.75 |
| Size | (1.25, 2.5, 1.25) | (2.5, 5.0, 2.5) | (3.75, 7.5, 3.75) |
|---|---|---|---|
| a F2 is correction factor to allow for the eccentricity of the ellipse, which equals unity for circular contact, obtained from the book.51 | |||
| A | s | 0.1 | 1/15 |
| B | 0.8 | 0.4 | 4/15 |
 |
2.0 | 2.0 | 2.0 |
| χ12η12 | 5.618 | 11.236 | 16.854 |
| Re (nm) | 1.25 | 2.5 | 3.75 |
| F2a | 0.99 | 0.99 | 0.99 |
| The number of Si atoms | 833 | 6498 | 22067 |
As observed from Table 2, both orientation parameter χ12η12 and overall equivalent radii Re increase with particle size, but their aspect ratios do not change.
Fig. 3 shows the size dependence of interaction potentials as a function of surface separation d between two parallel identical silicon nanoellipsoids. The results show that the LJ potentials decrease with the decrease of surface separation up to a minimum at d ≈ 0.25 nm, i.e., equilibrium separation where vdW attraction force equals Born repulsion force, and then increase sharply at small separation due to the dominant Born repulsion potential. Moreover, both vdW attraction and Born repulsion potentials (inset of Fig. 3) vary in a regular pattern. For a given surface separation, the magnitudes of vdW attraction or Born repulsion potential increase with particle size. For a given particle size, the magnitudes increase with the decrease of surface separation.
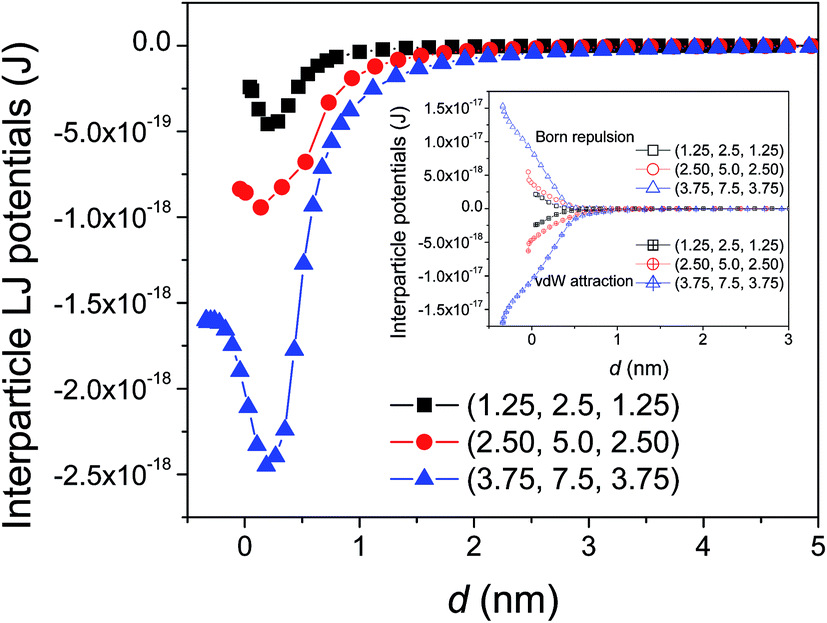 | ||
| Fig. 3 Interaction potentials as a function of the shortest surface separation d between silicon nanoellipsoids of different size but the same aspect ratio of 2. | ||
A further comparison is made between the MD simulated results and those calculated from the approximate equations of Hamaker approach for ellipsoids. The attractive and repulsive potential contributions between two ellipsoids can be approximately given by25
 | (3) |
 | (4) |
 is the orientation and relative position dependent part with a product of two terms.
is the orientation and relative position dependent part with a product of two terms.
Fig. 4 shows the ratios of the interaction forces obtained from the MD simulations to those predicted by Hamaker approach as a function of surface separation (d). The ratios of vdW attraction forces increase sharply with surface separation to a peak value around d ≈ 0.4 nm, followed by a sharp decrease and finally an asymptotic constant. In general, the MD simulated results are larger than those from Hamaker approach. Such deviation is usually ascribed to the surface effect, the neglect of the nature of discrete atomic structures,20,21,26,27 and the underestimated number density of atoms,52 and has been detailed before.32 The asymptotic ratio of vdW forces (kvdW) decreases with the increase of the product of χ12η12Re as shown in the inset of Fig. 4a. Such asymptotic ratio will be further related to aspect ratio  and surface roughness rms. For Born repulsion forces (Fig. 4b), comparing with the vdW attraction, there exist three differences: (i) there is no constant asymptotic ratio, (ii) the size-dependence is not clearly observed, and (iii) note that there exists a quite large difference between the MD simulated results and the predicted results from the simplified eqn (4). This is because Born repulsion decreases more rapidly with the surface separation and the approximation adopted in deriving the simplified eqn (4) results in such large deviation. Generally speaking, the force ratios increase sharply with surface separation d at small separation, followed by a gradual increase at larger separation except the smallest one whose ratio exists a minimum value around 0.85 nm.
and surface roughness rms. For Born repulsion forces (Fig. 4b), comparing with the vdW attraction, there exist three differences: (i) there is no constant asymptotic ratio, (ii) the size-dependence is not clearly observed, and (iii) note that there exists a quite large difference between the MD simulated results and the predicted results from the simplified eqn (4). This is because Born repulsion decreases more rapidly with the surface separation and the approximation adopted in deriving the simplified eqn (4) results in such large deviation. Generally speaking, the force ratios increase sharply with surface separation d at small separation, followed by a gradual increase at larger separation except the smallest one whose ratio exists a minimum value around 0.85 nm.
 | ||
Fig. 4 The ratios of interaction forces between identical silicon nanoellipsoids of different sizes but the same aspect ratio obtained from MD simulations to those from Hamaker approach: (a) vdW attraction and (b) Born repulsion. The inset shows the variation of asymptotic ratio kvdW as a function of 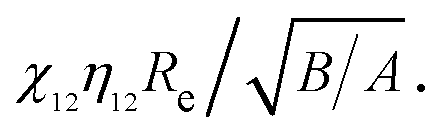 | ||
3.2. Aspect ratio
The effect of aspect ratio of silicon ellipsoids on interaction forces is also examined. The structural characteristics and parameters are determined in a similar way as in Section 3.1 using eqn (1) and (2) and shown in Tables 3 and 4. Table 4 shows that the orientation parameter χ12η12 and the overall equivalent radii Re do not change with the aspect ratio
| Apex | (1.25, 0, 0) | (2.5, 0, 0) | (3.75, 0, 0) |
|---|---|---|---|
| R′1 | 20.0 | 10.0 | 20/3 |
| R′′1 | 1.25 | 2.5 | 3.75 |

Fig. 5 shows the interaction potentials between two parallel nanoellipsoids as a function of surface separation d. The results show that the LJ potentials decrease with the decrease of surface separation to a minimum value at d ≈ 0.25 nm, and then increase sharply at small separation due to the dominant Born repulsion potentials. As shown in the inset of Fig. 5, for a given particle size, the magnitudes of both the vdW attraction and Born repulsion potentials increase with the decrease of surface separation.
 | ||
| Fig. 5 Interaction potentials between two parallel identical silicon nanoellipsoids with different aspect ratios as a function of the shortest surface separation d. | ||
As shown in Fig. 6a, the ratios of vdW attraction forces increase sharply with surface separation to a peak value around d = 0.4 nm, followed by a sharp decrease and finally an asymptotic constant. The asymptotic ratio of vdW forces (kvdW) decreases with the increase of the product of 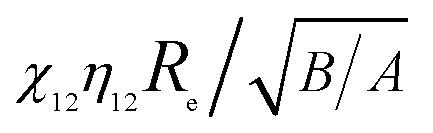 as shown in the inset of Fig. 6a. It is also related to surface roughness rms, given by
as shown in the inset of Fig. 6a. It is also related to surface roughness rms, given by
 | (5) |
 | ||
Fig. 6 The ratios of interaction forces between two silicon nanoellipsoids with different aspect ratios obtained from MD simulations to those from the equations: (a) vdW attraction; and (b) Born repulsion. The inset shows the variation of asymptotic ratio kvdW as a function of  and the open circles represent data points obtained from Fig. 4a. and the open circles represent data points obtained from Fig. 4a. | ||
As for Born repulsion forces, similar to the dependence on particle size, the same trend can be observed: in general, the force ratios increase sharply with surface separation d at small separation, followed by a gradual increase with surface separation except the case of (2.5, 5.0, 2.5) whose ratio exists a minimum value around d ≈ 0.85 nm (Fig. 6b).
The MD simulated results (Fig. 7) show that at d < 0.6 nm both vdW attraction and Born repulsion forces demonstrate a fluctuation. It is clear that the larger the particle size, the more pronounced the fluctuation. Therefore, the force obtained from MD simulation at d ≈ 0.6 nm is used as a reference when formulating interaction forces between ellipsoids at d < 0.6 nm. Such fluctuation can be attributed to the stepped atomic structures on ellipsoidal surface during their creation. The large particle size can lead to large contact area and hence relatively more pronounced fluctuations. The variation can be described by a second-order Fourier expansion, and vdW attraction forces can be represented by the following modified formulas,
 | (6) |
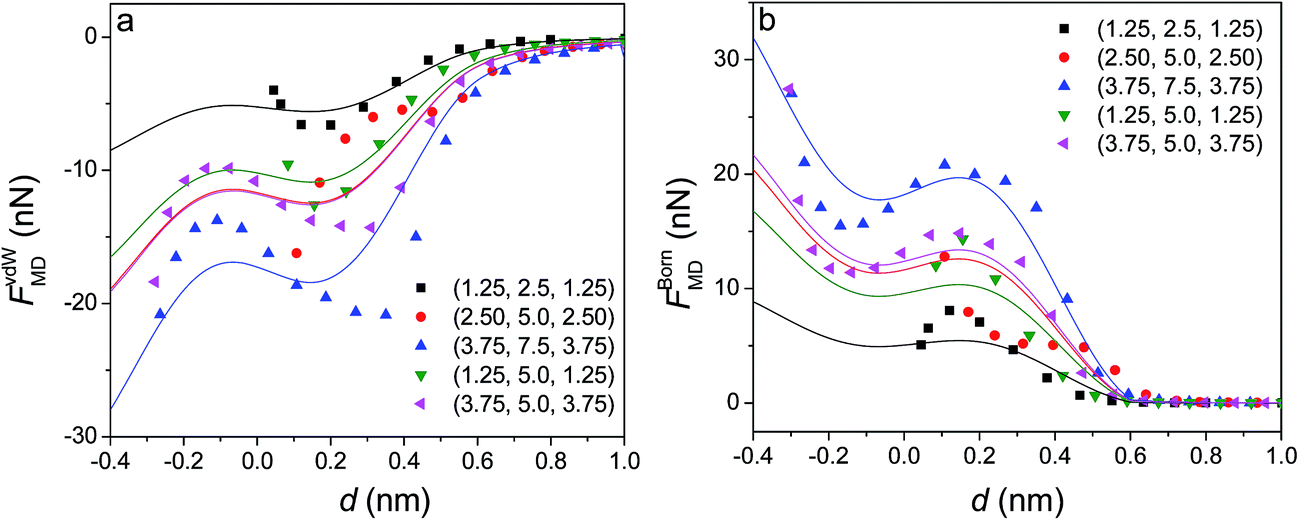 | ||
| Fig. 7 Interaction forces between two identical parallel silicon nanoellipsoids obtained from the MD simulations: (a), vdW attraction and (b), Born repulsion forces. The solid symbols represent the MD simulation results while the solid lines denote the proposed formulas. | ||
Similarly, the interaction Born repulsion forces can be described by the following modified formula. As shown in Fig. 7, the calculated forces from eqn (6) and (7) match reasonably with the MD simulated results.
 | (7) |
Of note, there exist some difficulties in direct comparison with experimental results in terms of measured vdW forces. The main barrier lies in the size of silicon nanoparticles. Due to the limiting computation efficiency, the size of nanoparticles in MD simulations is limited to below 5.0 nm in radius (in terms of equivalent nanosphere). Therefore, a compromise between size of modeled system and computation efficiency is made. In contrast, experimental studies on silicon nanospheres involve the particles ranging from 20 to 140 nm radius.53,54 What's more, most of them focus on contact forces. However, with the advancement of technology, this gap may be bridged in future. Nevertheless, the present simulated results closely resemble the experimental ones in a qualitative way. The interparticle LJ potential (equivalent to force), i.e., the total of vdW attraction and Born repulsion potentials, first decreases to a minimum with decreasing surface separation, then increases gradually at close separation and even changes sign and becomes repulsive force after contact deformation. This trend is similar to those measured in AFM experiments.55,56 In this work, in order to test the accuracy of COMPASS force field for silicon material, the Young's modulus of silicon bulk is separately measured by MD simulations, as obtained by the slope of the initial linear part of the established stress–strain curve (Fig. 8).33 The results show that Young's modulus along the (100) direction of crystalline silicon bulk is about 125.5 GPa, which is in an excellent agreement with the experimental value of 130 GPa,57 which can lend support to our current results.
 | ||
| Fig. 8 (a): simulation model of 6.0 × 4.0 × 4.0 nm3 silicon cluster used to measure Young's modulus of silicon bulk counterpart along the (100) direction; (b): sompressive stress–strain curve of silicon bulk. | ||
In addition to vdW attraction and Born repulsion forces, an additional mechanical contact force will arise upon particle contact deformation. Hertz theory is widely used to describe the contact force arisen from elastic deformation of solid surface,19 and later is extended to ellipsoidal particles by introducing an overall equivalent radius of curvature Re and considering the correction factor F2. Thus, the mechanical contact force is given by51
 | (8) |
The mechanical contact force Fc can also be measured by MD simulations and calculated by33
| Fc = Fn − FvdW − FBorn | (9) |
The calculated results of contact forces Fc using eqn (9) show that the first non-zero value of mechanical contact forces arise when surface separation is about d ≈ 0.2 nm. This value is consistent with the observed minima of interparticle LJ potentials from Fig. 3 and 5. The minimum point of LJ potential (i.e., the zero point of LJ force) corresponds to the point where the occurrence of mechanical contact force initiates, confirming that due to the intermolecular repulsive forces, the surface atoms are prone to be subject to “deformation” and hence the mechanical contact force arises when two surfaces are less than equilibrium separation distance apart (Fig. S-3, ESI†). Fig. 9 shows the normalized mechanical contact forces Fc/(4 × 21.5Re2E*/3F21.5) between two identical parallel silicon nanoellipsoids as a function of strain δn/(2Re). The MD simulated results agree reasonably with Hertz predictions in particular at small deformation. The deviation at large deformation can be ascribed to the effect of high internal pressure or the creation of new phases.33
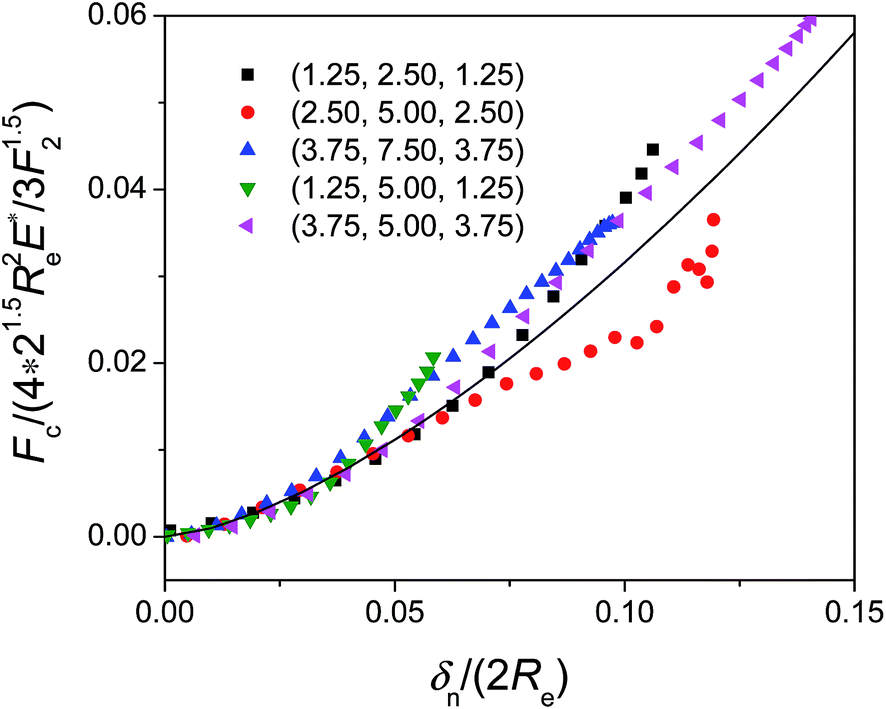 | ||
| Fig. 9 Normalized mechanical contact forces between two identical parallel silicon nanoellipsoids obtained from MD simulations as a function of δn/(2Re) at Vr,0 = 400 m s−1 δn = 0 corresponds to the starting point of the first non-zero value of the mechanical contact force. The solid line represents the prediction by Hertz model. | ||
3.3. Configuration dependence
For non-spherical particles, different configuration (i.e., relative orientation between two ellipsoids) can significantly affect the interaction forces. Thus, four typical configurations of two identical silicon ellipsoidal nanoparticles are examined (Fig. 10). The interaction forces measured from the MD simulations are compared to those from Hamaker approach and Hertz model. | ||
| Fig. 10 Illustrations of four typical configurations of two identical silicon nanoellipsoids (2.5 nm, 5.0 nm, 2.5 nm): (a) parallel (∥), (b) crossed (+), (c) end-to-side (⊥) and (d) end-to-end (--). | ||
The principle radii of curvatures at each apex are calculated in a similar way as done in Sections 3.1 and 3.2 (Table 5) and structural parameters including A, B,  , χ12η12, Re are obtained (Table 6). The results show that the asymptotic ratio of the vdW forces (kvdW) still varies with the product of
, χ12η12, Re are obtained (Table 6). The results show that the asymptotic ratio of the vdW forces (kvdW) still varies with the product of  in a regular pattern (Fig. 12a), though
in a regular pattern (Fig. 12a), though 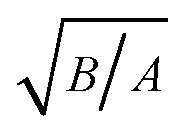 , χ12η12, Re all vary with different configurations.
, χ12η12, Re all vary with different configurations.
| Apex | (2.5, 0, 0) | (0, 5.0, 0) | (0, 0, 2.5) |
|---|---|---|---|
| R′1 | 2.5 | 1.25 | 10 |
| R′′1 | 10 | 1.25 | 2.5 |

 | ||
| Fig. 11 Interaction potentials as a function of the shortest surface separation d between two identical silicon nanoellipsoids with different configurations at Vr,0 = 400 m s−1. | ||
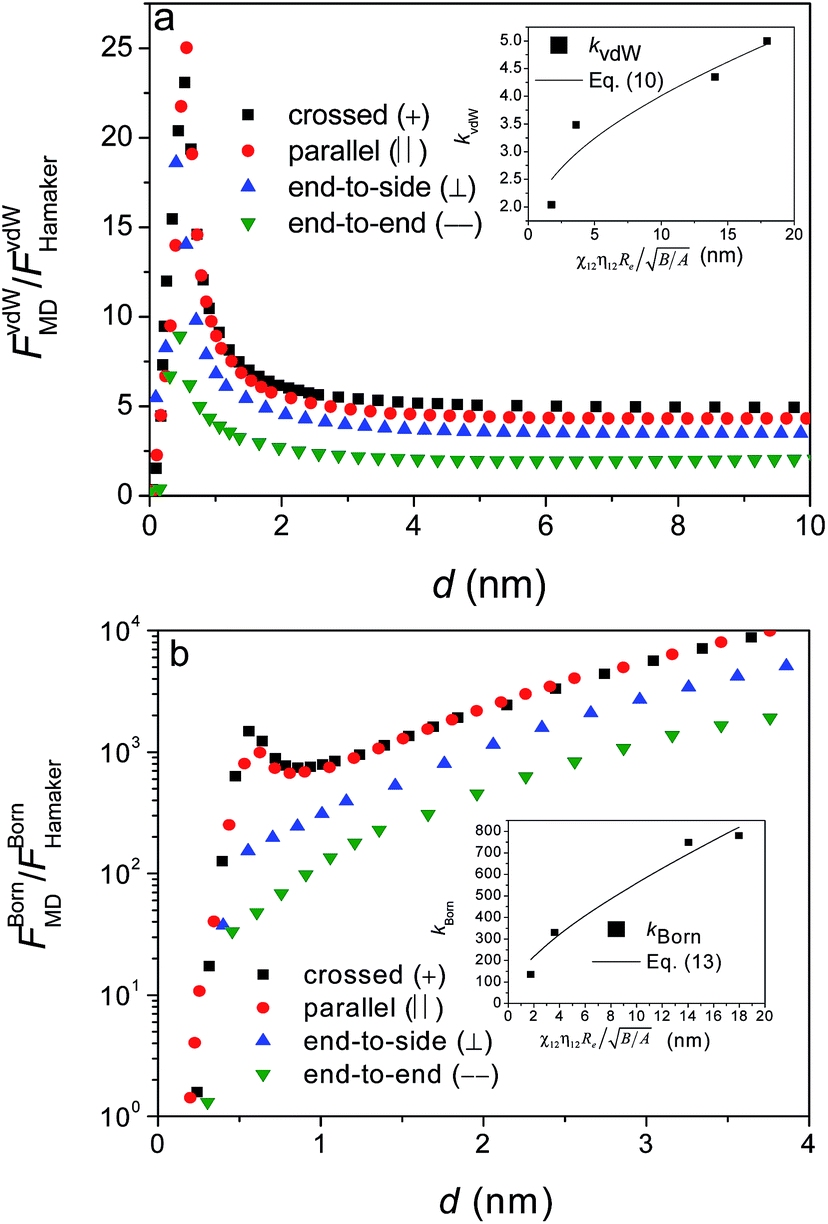 | ||
Fig. 12 The ratios of interaction forces between two identical silicon nanoellipsoids of different configurations obtained from MD simulations to those from Hamaker approach: (a) vdW attraction; and (b) Born repulsion. The insets show the dependence of kvdW and kBorn on  . . | ||
Fig. 11 shows the dependence of the interaction potentials on the configuration. It can be observed that LJ potentials decrease with decreasing surface separation to a minimum at d = 0.25–0.30 nm, and then increase sharply at small separation because of the dominant Born repulsion potentials. As shown in the inset of Fig. 11, both the vdW attraction and Born repulsion potentials vary in a regular manner: for a given separation, the magnitudes of both the vdW interaction potentials and Born repulsion potentials increase in the order of crossed, parallel, end-to-side and end-to-end configurations.
The ratios of vdW attraction forces (Fig. 12a) increase sharply with surface separation to a peak value around d ≈ 0.4 nm, followed by a quick decrease and finally an asymptotic constant. However, contrary to the trends observed from the dependence of particle size and aspect ratio, the asymptotic ratio of the forces (kvdW) increases (but does not decrease any more as shown in Fig. 4a and 6a) with the product of 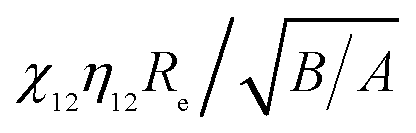 in order of crossed, parallel, end-to-side and end-to-end (inset of Fig. 11a). Because of this, interaction forces under parallel configurations are separately considered and two relatively accurate formulas have been proposed to describe vdW attraction and Born repulsion forces, i.e., eqn (6) and (7) in above Subsection 3.2.
in order of crossed, parallel, end-to-side and end-to-end (inset of Fig. 11a). Because of this, interaction forces under parallel configurations are separately considered and two relatively accurate formulas have been proposed to describe vdW attraction and Born repulsion forces, i.e., eqn (6) and (7) in above Subsection 3.2.
As for the cases under different configurations, the asymptotic ratios of the vdW attraction forces are related to surface roughness rms, given by
 | (10) |
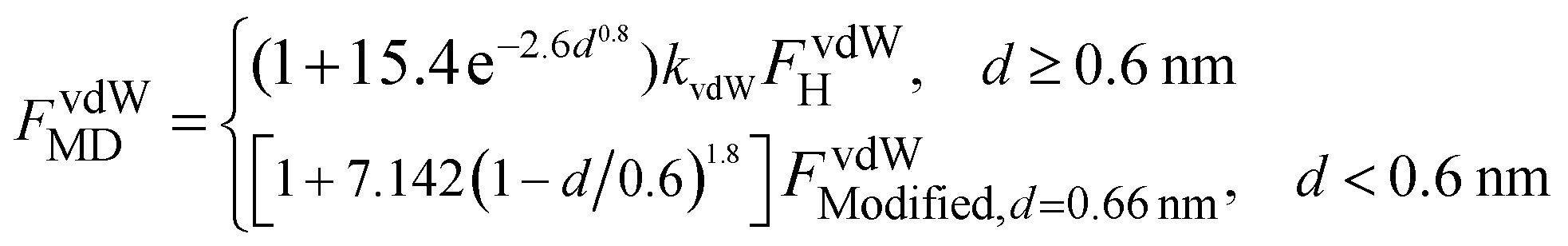 | (11) |
Note that FvdWModified,d=0.6 nm is the vdW attraction force calculated from the first formula in eqn (11) at the surface separation of d = 0.6 nm.
For the Born repulsion forces (Fig. 12b), the ratios for the cases of crossed and parallel first increase with surface separation, then decrease sharply. But finally the ratios for all the cases increase monotonically with surface separation d at d ≥ 1.0 nm and display an obvious dependence on configurations. The ratio kBorn at d ≈ 1.0 nm is used to formulate the Born repulsion force, giving rise to the following formulas,
 | (12) |
 and can be calculated by
and can be calculated by
 | (13) |
Note that FBornModified,d=1.0 nm is the Born repulsion force calculated from the first formula in eqn (12) at the surface separation of d = 1.0 nm.
As shown in Fig. 13, the calculated forces can reasonably match the MD simulated results. For a given silicon nanoellipsoid, the periodic variation of vdW attraction and Born repulsion between two identical ellipsoids under different configurations becomes vague, which is different from the results in Fig. 7. Therefore, different forms have been taken when formulating interaction forces. Such difference can be ascribed to the fact that under parallel configuration, the contact area is relatively higher than those in other configurations. With increase in deformation, the contact area under any configuration can increase and hence the periodic variation may happen. However, at large deformation, the mechanical contact force becomes increasingly important and the role of intermolecular forces become less important, therefore our attention is mainly paid to intermolecular forces at small deformation.
 | ||
| Fig. 13 The interaction forces between two silicon nanoellipsoids obtained from atomistic MD simulations and the proposed formulas: (a) vdW attraction and (b) Born repulsion. At d < 0.6 or 1.0 nm, the ratio of FvdWMD/FvdWModified,d=0.6 nm or FBornMD/FBornModified,d=1.0 nm is instead used, respectively. | ||
The mechanical contact forces Fc between silicon nanoellipsoids under different configurations are calculated by MD simulation and eqn (9), respectively. The normalized mechanical contact force Fc/(4 × 21.5Re2E*/3F21.5) between silicon nanoellipsoids under different configurations (Fig. 14) are comparable to those predicted by Hertz theory but there is a small deviation at large deformation, indicating that Hertz model still holds to describe mechanical contact force between nanoellipsoids at small deformation.
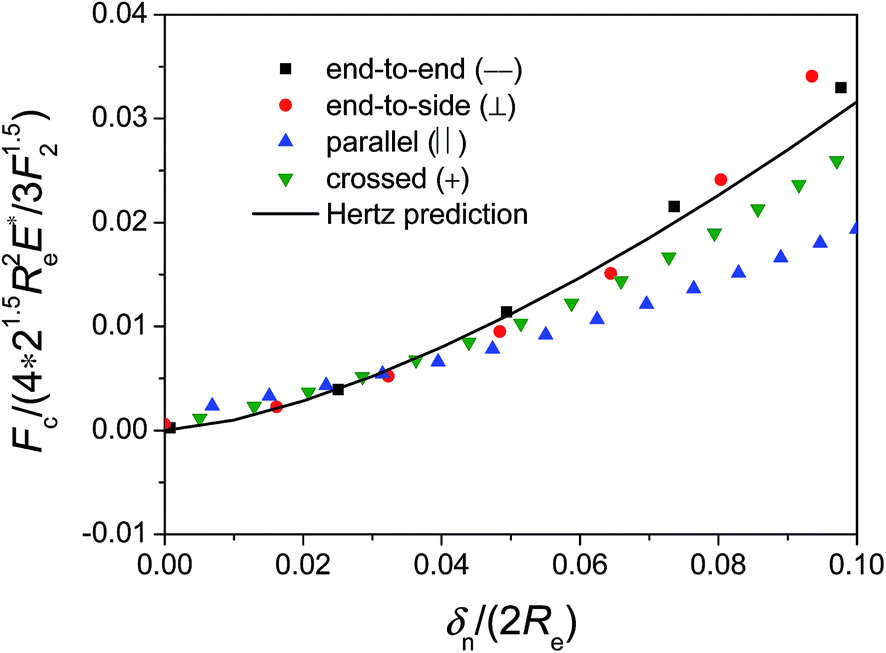 | ||
| Fig. 14 Normalized mechanical contact force as a function of δn/(2Re) obtained from the MD simulations between two silicon nanoellipsoids under different configurations at Vr,0 = 400 m s−1. Solid line represents Hertz prediction. | ||
It is worth mentioning that when the particle size comes down to such ultrasmall size below 10 nm, the inter-nanoparticle forces are sensitive to a variety of subtle factors: the random motions of surface atoms, the atomic roughness arisen from amorphous or stepped (commensurate or incommensurate) interfaces. However, from the point of practical applications, it is still valuable to provide some insights into the self-assembly, design of nanodevices and can be used as relatively accurate approximation since one rigorous theory is difficult to achieve at the atomic level.
4. Conclusions
In this work, the validity of the adapted continuum models pertinent to ellipsoidal particles has been tested at the nanoscale. And the interaction forces including vdW attraction, Born repulsion and mechanical contact forces between silicon nanoellipsoids have been studied by MD simulations and their dependences on particle size, aspect ratio and configurations are explored. The results show that the adapted continuum Hamaker model breaks down at the nanoscale, which is ascribed to the approximations apart from the surface effects, the neglect of atomic discrete structure and the underestimated number density of atoms. For vdW attraction and Born repulsion forces, the MD simulated results are larger than those calculated from Hamaker approach. And the ratios of interaction forces are complicated and dependent on surface separation, overall equivalent radius of curvature, aspect ratio and configuration. In particular, under parallel configuration, both the vdW attraction and Born repulsion upon contact deformation between two nanoellipsoids show a pronounced periodic variation that can be described by a second-order Fourier expansion, and two relatively accurate equations have been proposed for vdW attraction and Born repulsion forces. In contrast, under different configuration, the periodic variation of vdW attraction and Born repulsion becomes vague and another two equations have been separately proposed for vdW attraction and Born repulsion forces between two nanoellipsoids. Moreover, the mechanical contact forces between nanoellipsoids can be largely described by the continuum Hertz model at low compression.Finally, the advancement of understanding interaction forces between nanoellipsoids may shed some light on not only practical applications, such as assembly of non-spherical particles, but also the understanding of packing, flow and other particle behaviors using the cost-effective discrete element simulation in future.
Acknowledgements
The authors gratefully acknowledge the financial funding by the Australian Research Council (ARC).Notes and references
- P. Schiller, S. Kruger, M. Wahab and H. J. Mogel, Langmuir, 2011, 27, 10429–10437 CrossRef CAS PubMed.
- H. Imura and K. Okano, J. Chem. Phys., 1973, 58, 2763–2776 CrossRef PubMed.
- J. C. Loudet, A. M. Alsayed, J. Zhang and A. G. Yodh, Phys. Rev. Lett., 2005, 94, 018301 CrossRef CAS.
- P. J. Yunker, T. Still, M. A. Lohr and A. G. Yodh, Nature, 2011, 476, 308–311 CrossRef CAS PubMed.
- J. Park and J. Moon, Langmuir, 2006, 22, 3506–3513 CrossRef CAS PubMed.
- V. Dugas, J. Broutin and E. Souteyrand, Langmuir, 2005, 21, 9130–9136 CrossRef CAS PubMed.
- B. J. de Gans, P. C. Duineveld and U. S. Schubert, Adv. Mater., 2004, 16, 203–213 CrossRef CAS PubMed.
- W. F. Sun, G. H. Chen and L. L. Zheng, Scr. Mater., 2008, 59, 1031–1034 CrossRef CAS PubMed.
- A. P. Alivisatos, Science, 1996, 271, 933–937 CAS.
- A. B. Yu, X. Z. An, R. P. Zou, R. Y. Yang and K. Kendall, Phys. Rev. Lett., 2006, 97, 265501 CrossRef CAS.
- Q. Li, U. Jonas, X. S. Zhao and M. Kapp, Asia-Pac. J. Chem. Eng., 2008, 3, 255–268 CrossRef CAS PubMed.
- R. Nidetz and J. Kim, Nanotechnology, 2012, 23, 045602 CrossRef PubMed.
- N. R. Jana, Angew. Chem., Int. Ed., 2004, 43, 1536–1540 CrossRef CAS PubMed.
- X. C. Jiang, Q. H. Zeng, C. Y. Chen and A. B. Yu, J. Mater. Chem., 2011, 21, 16797–16805 RSC.
- L. S. Cheng and D. P. Cao, Langmuir, 2009, 25, 2749–2756 CrossRef CAS PubMed.
- C. B. Xu and J. Zhu, Chem. Eng. Sci., 2005, 60, 6529–6541 CrossRef CAS PubMed.
- H. P. Zhu, Z. Y. Zhou, R. Y. Yang and A. B. Yu, Chem. Eng. Sci., 2007, 62, 3378–3396 CrossRef CAS PubMed.
- H. C. Hamaker, Physica, 1937, 4, 1058–1072 CrossRef CAS.
- H. Hertz, J. Reine Angew. Math., 1882, 92, 156–171 Search PubMed.
- B. Q. Luan and M. O. Robbins, Nature, 2005, 435, 929–932 CrossRef CAS PubMed.
- K. J. M. Bishop, C. E. Wilmer, S. Soh and B. A. Grzybowski, Small, 2009, 5, 1600–1630 CrossRef CAS PubMed.
- D. L. Feke, N. D. Prabhu, J. A. Mann and J. A. Mann, J. Phys. Chem., 1984, 88, 5735–5739 CrossRef CAS.
- J. G. Gay and B. J. Berne, J. Chem. Phys., 1981, 74, 3316–3319 CrossRef CAS PubMed.
- J. W. Perram, J. Rasmussen, E. Praestgaard and J. L. Lebowitz, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1996, 54, 6565–6572 CrossRef CAS.
- R. Everaers and M. R. Ejtehadi, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2003, 67, 041710 CrossRef CAS.
- L. A. Girifalco, M. Hodak and R. S. Lee, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, 13104–13110 CrossRef CAS.
- V. A. Parsegian, van der Waals forces: a handbook for biologist, chemists, engineers, and physicist, Cambridge University Press, Cambridge, 2005 Search PubMed.
- L. O. Heim, J. Blum, M. Preuss and H. J. Butt, Phys. Rev. Lett., 1999, 83, 3328–3331 CrossRef CAS.
- X. Ling, H. J. Butt and M. Kappl, Langmuir, 2007, 23, 8392–8399 CrossRef CAS PubMed.
- B. J. Yu, H. S. Dong, L. M. Qian, Y. F. Chen, J. X. Yu and Z. R. Zhou, Nanotechnology, 2009, 20, 465303 CrossRef PubMed.
- Q. H. Zeng, A. B. Yu and G. Q. Lu, Ind. Eng. Chem. Res., 2010, 49, 12793–12797 CrossRef CAS.
- W. F. Sun, Q. H. Zeng and A. B. Yu, Langmuir, 2013, 29, 2175–2184 CrossRef CAS PubMed.
- W. F. Sun, Q. H. Zeng, A. B. Yu and K. Kendall, Langmuir, 2013, 29, 7825–7837 CrossRef CAS PubMed.
- J. A. Elliott, Y. Shibuta, H. Amara, C. Bichara and E. C. Neyts, Nanoscale, 2013, 5, 6662–6676 RSC.
- L. Xu, T. B. Ma, Y. Z. Hu and H. Wang, Nanotechnology, 2011, 22, 285708 CrossRef PubMed.
- W. M. Huang, W. F. Sun, G. H. Chen and L. Tan, Adv. Eng. Mater., 2014 DOI:10.1002/adem.201400143.
- W. F. Sun, Q. H. Zeng and A. B. Yu, Chem. Eng. Sci., 2014 DOI:10.1016/j.ces.2014.07.023.
- J. P. Alper, A. Gutes, C. Carraro and R. Maboudian, Nanoscale, 2013, 5, 4114–4118 RSC.
- L. Dai, L. Zhang, L. X. Dong, W. Z. Shen, X. B. Zhang, Z. Z. Ye and B. J. Nelson, Nanoscale, 2011, 3, 4301–4306 RSC.
- M. Zhu, L. Zhou, B. Li, M. K. Dawood, G. Wan, C. Q. Lai, H. Cheng, K. C. Leong, R. Rajagopalan, H. P. Too and W. K. Choi, Nanoscale, 2011, 3, 2723–2729 RSC.
- H. Sun, J. Phys. Chem. B, 1998, 102, 7338–7364 CrossRef CAS.
- H. Sun and D. Rigby, Spectrochim. Acta, Part A, 1997, 53, 1301–1323 CrossRef.
- W. F. Sun, Y. C. Li, W. Xu and Y. W. Mai, RSC Adv., 2014, 4, 34500–34509 RSC.
- W. F. Sun, Nanoscale, 2013, 5, 12658–12669 RSC.
- H. Y. Kim, J. O. Sofo, D. Velegol, M. W. Cole and A. A. Lucas, J. Chem. Phys., 2006, 124, 74504 CrossRef PubMed.
- G. Han, Z. G. Chen, C. H. Sun, L. Yang, L. N. Cheng, Z. F. Li, W. Lu, Z. M. Gibbs, G. J. Snyder, K. Jack, J. Drennan and J. Zou, CrystEngComm, 2014, 16, 393–398 RSC.
- K. H. Lin, C. H. Sun, S. P. Ju and S. C. Smith, Int. J. Hydrogen Energy, 2013, 38, 6283–6287 CrossRef CAS PubMed.
- C. H. Sun and D. J. Searles, J. Phys. Chem. C, 2012, 116, 26222–26226 CAS.
- C. H. Sun, A. J. Du, X. D. Yao and S. C. Smith, J. Phys. Chem. C, 2011, 115, 12580–12585 CAS.
- A. K. Rappe, C. J. Casewit, K. S. Colwell, W. A. Goddard and W. M. Skiff, J. Am. Chem. Soc., 1992, 114, 10024–10035 CrossRef CAS.
- K. L. Johnson, Contact mechanics, Cambridge University Press, Cambridge, 2rd edn, 1985 Search PubMed.
- W. F. Sun, Phys. Chem. Chem. Phys., 2014, 16, 5846–5854 RSC.
- W. W. Gerberich, W. M. Mook, C. R. Perrey, C. B. Carter, M. I. Baskes, R. Mukherjee, A. Gidwani, J. Heberlein, P. H. McMurry and S. L. Girshick, J. Mech. Phys. Solids, 2003, 51, 979–992 CrossRef CAS.
- W. M. Mook, J. D. Nowak, C. R. Perrey, C. B. Carter, R. Mukherjee, S. L. Girshick, P. H. McMurry and W. W. Gerberich, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 214112 CrossRef.
- L. F. Hakim, J. H. Blackson and A. W. Weimer, Chem. Eng. Sci., 2007, 62, 6199–6211 CrossRef CAS PubMed.
- G. Friedbacher, E. Bouveresse, G. Fuchs, M. Grasserbauer, D. Schwarzbach, R. Haubner and B. Lux, Appl. Surf. Sci., 1995, 84, 133–143 CrossRef CAS.
- M. A. Hopcroft, W. D. Nix and T. W. Kenny, J. Microelectromech. Syst., 2010, 19, 229–238 CrossRef CAS.
- Q. J. Zheng, Z. Y. Zhou and A. B. Yu, Powder Technol., 2013, 248, 25–33 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra06809b |
| ‡ Current address. School of Aerospace, Mechanical and Mechatronic Engineering, The University of Sydney, NSW 2006, Australia. |
| This journal is © The Royal Society of Chemistry 2014 |