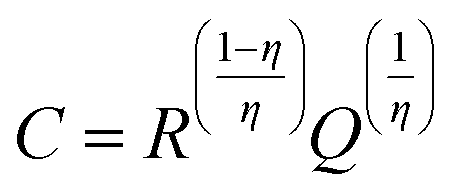DOI:
10.1039/C4RA06678B
(Paper)
RSC Adv., 2014,
4, 35717-35725
Nitrogen-doped carbon coated SiO nanoparticles Co-modified with nitrogen-doped graphene as a superior anode material for lithium-ion batteries†
Received
5th July 2014
, Accepted 4th August 2014
First published on 5th August 2014
Abstract
One great challenge in the development of lithium-ion batteries is to simultaneously achieve superior reversible specific capacity, cyclic life and rate capability. In this work, nitrogen-doped carbon coated Silicon monoxide nanoparticles further co-modified with nitrogen-doped graphene were developed. The nanostructure of Silicon monoxide was designed and realized by introducing the highly efficient attritor mill technique, which assisted in forming optimized morphology and particle size distributions of the precursor. The nitrogen-doped carbon coating process was achieved by a simple surface coating technique using a carbon and nitrogen containing ionic liquid as a precursor, and the nitrogen-doped graphene was prepared by a facile, catalyst-free thermal annealing approach using low-cost industrial material melamine as the nitrogen source. XRD, XPS, RAMAN, FESEM, EDAX, TEM, HRTEM, AFM, BET, elemental analysis, electrical conductivity measurement and electrochemical methods were used to characterize the material's properties. The results showed that the reduced active particle size, coupled with the co-modification of the NC coating layer and NG network could effectively construct a 3D conducting network through a 3D “sheet-web” mode. As a result, the composite material showed exceptional high reversible specific capacity, ultra long cyclic life and superior high-rate capability. The present strategy opens up the possibility for integrating other anode materials with large volume variations and low electrical conductivities into current lithium-ion battery manufacture technology.
Introduction
The development of high energy, high-power, long-life and low-cost lithium-ion batteries (LIBs) is of great technological importance for use in mobile devices, electric vehicles (EVs), hybrid electric vehicles (HEVs) and renewable energy storage.1–3 To meet these requirements, the electrode materials of LIBs must have high-performance electrochemical properties, such as high stability, high conductivity and high Li+ diffusion rate. Compared with the graphite anode, which has a limited theoretical specific capacity of 372 mA h g−1, silicon monoxide (SiO) has been proposed as one of most promising anode candidates for next-generation LIBs, owing to its significant high theoretical capacity was reported to be 2400 mA h g−1,4 and low charge–discharge potential. However, a large volume change of SiO particles upon electrochemical cycling (close to 200% of the initial volume)5 and the intrinsically low electric conductivity (6.7 × 10−4 S cm−1)6 significantly restrict the high rate capability of fast charging and discharging of active material, resulting in a poor power performance and thus inhibiting the commercialization of the SiO-based materials in LIBs. The tremendous efforts have been made to overcome these drawbacks by reducing the particle size to shorten the Li+ diffusion path,7–9 doping foreign ions or atoms to enhance Li+ diffusion velocity,10–12 changing to more effective binders to suppress swelling of the composite electrode13 and modifying with buffer materials (which is in general carbon) to increase electronic conductivity.9,14–16 Various carbon sources can be used, including amorphous carbon and graphene: firstly, carbon-coating is considered as one of the most effective and conventional ways to improve the electrochemical properties. As compared to the conventional carbon-coated counterpart, the N-doped carbon (NC) coating layers significantly improve the interfacial stability and interfacial electric conductivity, resulting from a remarkable electron transfer from the surface of active material to the coating layers, yielding desirable electrochemical properties for energy storage applications.12,17–19 Secondly, as a one-atom-thick two-dimensional (2D) carbon material, graphene is expected to be a good candidate as a high-power and high-energy electrode material due to its intrinsically superior electrical conductivity, excellent mechanical flexibility, remarkable thermal conductivity and high surface area, as well as the open and flexible porous structure of graphene powders.20–22 Recent reports demonstrated that nitrogen-doped (N-doped) graphene (NG) shows more excellent electrochemistry performance for LIBs as compared to pristine graphene (PG).23–25 The significant improvement is due to the fact that the N atoms introduction in the graphene plane leads to an increase in the number of defect sites and vacancies as Li+ active sites on the surface of NG sheet.26,27 Based on these results, we expect that NG will be used as a template for synthesis of active electrode materials. However, most previous trials merely focused on one of the above strategies, leading to a limited improvement in the performance. In our previous work,28 we reported a study on SiOx with the co-modification strategy using a carbon coating layer and reduced graphene oxide (RGO) network that exhibited very high reversible capacity (over 2000 mA h g−1), excellent cyclic stability and high-rate capability. However, synthesis procedure for the SiOx/C is discontinuous and complicated. In this article, a high efficient attritor mill (HEAM) technique is designed to deal with bulk precursors (commercially available SiO powders) and achieve particle size diminishment, thus the attritor-milled SiO (ASiO) nanoparticles were obtained. The reason for the selection of the HEAM is its advantages of high yield and high efficiency. Namely, compared with a solution chemistry method,28 the attritor mill process is still an economical and simple route without the expensive investment and complicated facilities. In addition, ionic liquids (IL) were used as precursors to obtain N-doped graphitized carbon.17–19 Compared with conventional solid carbon precursors, IL can penetrate into porous materials easily, because of their fluidic properties. We have applied a simple surface coating technique using a carbon and N-containing IL as a precursor to deposit a thin layer of N-doped carbon on a ASiO electrode, denoted as NC–ASiO. Finally, elastic NG networks are then introduced for dispersing, embedding, and electrically wiring the NC–ASiO core–shell nanoparticles, thus forming a three dimensional (3D) nanocomposite material, denoted as ASiO/(NC + NG). In this co-modification strategy, the NC shell can act as a buffer that minimizes volume changes and direct contacts between ASiO nanoparticles and electrolyte, facilitating the formation of a stable solid electrolyte interphase (SEI) film which leads to outstanding cycling performance. Meanwhile, NC shell is regarded as a conducting bridge between ASiO nanoparticles and NG in the nanocomposites. The NG is an excellent capacitor material due to its distinctive properties23–27 which can instantly store and release a great quantity of charges. However, to the best of our knowledge, there were fewer reports on the ASiO/(NC + NG) nanocomposites as an anode material for LIBs so far. Herein, a tiny SiO nanoparticles co-modified by NC coating layer and NG network was synthesized. The as-prepared ASiO/(NC + NG) nanocomposites may exhibit synergistic properties and display superior electrochemical performance with large reversible specific capacity, excellent cycling performance and high rate capability, when used as anode for rechargeable LIBs. For comparison purpose, undoped carbon-coated ASiO (denoted as C–SiO), PG and NC–ASiO nanoparticles wrapped by PG (denoted as ASiO/(NC + PG)) were also prepared and investigated. In addition, although it seems still under debate, the reaction mechanism of SiO with Li has been reported by several groups.68–71 In our opinion, a model for the reaction of Li insertion into SiO was suggested. Employing the revised random mixture model for SiO,71 which was comprised of metallic Si clusters and SiO2 domain, the final products consisted of Li–Si alloy (LixSi), Li2O and Li4SiO4.
Experiment
Materials synthesis
Scheme 1 illustrates the synthesis procedure for the ASiO/(NC + NG) nanocomposites. The commercially available pristine SiO powders (Aldrich) (denoted as PSiO) with an average particle size of 40 μm was treated with high efficient attritor mill (HEAM) technique in a hardened steel vial with zirconia balls in ethanol, using a 01-HD/HDDM Lab Attritor with intensive rotation speed at 2500 rpm for 2 h. In order to keep a low processing temperature, after 30 min HEAM, there is a pause for 20 min, and then repeated this whole process for 3 times. The resultant dark brown precipitate was dried at 60 °C under vacuum conditions, then the ASiO nanoparticles was obtained. For the synthesis of NC–ASiO nanoparticles, the IL 1-ethyl-3-methylimidazolium dicyanamide (EMIm-dca) was chosen as the precursor, which is composed of only C, N, and H elements. The NC–ASiO nanoparticles were prepared by a carbonization reaction modifying a procedure reported by Zhao.19 In a typical synthesis procedure: 1 g of ASiO nanoparticles was first mixed with 300 μl of EMIm-dca IL (C-Tri, Korea), and the mixture was then heat-treated to 600 °C under Ar atmosphere. C–SiO nanoparticles were prepared by the same pyrolysis process but used sucrose as a carbon precursor. Graphene Oxide (GO) was prepared from purified natural graphite according to Hummers method as previous reports.29,30 NG was prepared by a facile and catalyst-free approach reported by Sheng31 as follows: GO powder and melamine were ground together in a mortar using pestle, and the mixture was then placed in the center of a corundum tube with a flow of argon. When the center of the furnace reached 800 °C, the mixture was annealed for 1 h. After that, the sample was cooled to room temperature under Ar ambient. The PG was synthesized from GO using a similar procedure but without melamine. The ASiO/(NC + NG) nanocomposites was prepared through dispersing the two materials in ethanol solution. Firstly, 10 mg of NG was ultrasonicated with 20 ml of ethanol for 1 h. Secondly, 40 mg of the prepared NC–SiO nanoparticles powder was added into the solution under magnetic stirring for 1 h to obtain a homogeneous suspension, then slowly drying at 60 °C. During the solvent vaporization, the NC–SiO nanoparticles deposited gradually and embedded into the NG network through van der Waals. The resultant solid products were collected by filtration and dried in vacuum at 80 °C for 12 h. For comparison, the NC–SiO nanoparticles modified with PG were also prepared by the same process with the addition of PG (noted as ASiO/(NC + PG)).
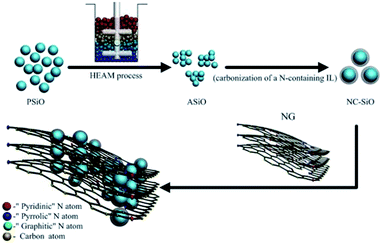 |
| | Scheme 1 Illustration of the preparation process and microstructure characteristics of the ASiO/(NC + NG) nanocomposite. | |
Materials characterization
X-ray diffraction (XRD) patterns of the as-prepared materials were collected on a D/max-γB X-ray diffractometer (Rigaku, Japan) using Cu Kα radiation (λ = 1.54178 Å). X-ray photoemission spectroscopy (XPS) measurements were performed on a spectrophotometer (PHI Quantera SXM) using the monochromatic AlKa radiation (1486.6 eV) to evaluate the elemental compositions and chemical status of the samples. In the XPS analysis, we used the carbon C 1s peak at 284.6 eV as a reference for charge correction. Raman spectra were recorded on a LabRAM HR800 Raman microprobe (HORIBA JY) with 632.8 nm laser excitation. The elemental composition was obtained from the CHN elemental analysis (Yanako MT2 CHN Corder, Japan). The morphology and microstructure of samples were characterized by field emission scanning electron microscopy (FESEM) (Hitachi, S-4800), transmission electron microscopy (TEM) (Hitachi, S-7650) and high resolution transmission electron microscopy (HRTEM) (JEM-2100). For atom force microscopy (AFM) measurement, the samples were coated on Si surface and AFM studies were performed using a Digital Instruments Dimension 3100 microscope in the tapping mode. The electrical conductivities of samples were measured at room temperature using a four-probe conductivity test metre (SB120; San Feng). The nitrogen physisorption at 77 K was performed using a Micromeritics ASAP 2020 surface area and porosity analyzer.
Electrochemical measurement
Electrochemical measurements were performed with coin-type cells (CR2016) assembled in an argon-filled glovebox (MBRAUN). The working electrode was fabricated by dispersing active materials (80 wt%), acetylene black (10 wt%), and a sodium carboxymethyl cellulose (CMC) binder (10 wt%) in de-ionized water. The solution was held at room temperature and stirred with a magnetic stirring apparatus. After stirring for 4 hours, a homogeneous slurry was obtained. The slurry was plastered on an Cu foil and then dried at 100 °C for 10 h in a vacuum oven. A Li foil and a polypropylene microporous film (Celgard 2400) were used as the counter electrode and separator, respectively. The electrolyte used was EC/DMC/DEC-based (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 by weight) electrolytes containing 1 M of LiPF6. The cells were charged and discharged over a voltage range of 0.01–1.5 V (vs. Li/Li+) at different current rates which were independent of the testing procedure using a Battery Testing System (Neware, China). Electrochemical impedance spectroscopy (EIS) and cyclic voltammetry (CV) were performed on an electrochemical workstation (PARSTAT 2273, Princeton Applied Research, U.S.A.). CV was carried out at a scanning rate of 0.1 mV s−1 between 0.01–3.0 V. EIS measurements were performed in the frequency range from 100 kHz to 10 mHz and recorded using two-electrode cells. The parameters of the equivalent circuit were calculated and analyzed by computer simulations using the ZSimpWin software. The weight of ASiO/(NC + NG) nanocomposites in the working electrode was used to estimate the specific discharge capacity of the battery, which was expressed in mA h g−1 of ASiO/(NC + NG). Reproducibility of the electrochemical data was confirmed by repeating the experiments with at least another electrode of the same sample. On the basis of the equation described below, we could calculate a theoretical capacity (Q) of the hypothetical mixture of ASiO/(NC + NG), as follows:
:1:1 by weight) electrolytes containing 1 M of LiPF6. The cells were charged and discharged over a voltage range of 0.01–1.5 V (vs. Li/Li+) at different current rates which were independent of the testing procedure using a Battery Testing System (Neware, China). Electrochemical impedance spectroscopy (EIS) and cyclic voltammetry (CV) were performed on an electrochemical workstation (PARSTAT 2273, Princeton Applied Research, U.S.A.). CV was carried out at a scanning rate of 0.1 mV s−1 between 0.01–3.0 V. EIS measurements were performed in the frequency range from 100 kHz to 10 mHz and recorded using two-electrode cells. The parameters of the equivalent circuit were calculated and analyzed by computer simulations using the ZSimpWin software. The weight of ASiO/(NC + NG) nanocomposites in the working electrode was used to estimate the specific discharge capacity of the battery, which was expressed in mA h g−1 of ASiO/(NC + NG). Reproducibility of the electrochemical data was confirmed by repeating the experiments with at least another electrode of the same sample. On the basis of the equation described below, we could calculate a theoretical capacity (Q) of the hypothetical mixture of ASiO/(NC + NG), as follows:
| Qtheoretical = QSiO × mass percentage of SiO + Qgraphite × mass percentage of graphite = 2400 × 60.39% + 372 × 30.47% = 1563 mA h g−1 |
The theoretical capacity of SiO was calculated on the basis of theoretical capacity of SiO previously reported.7 The calculated method of theoretical capacity was also applied to ASiO/(NC + PG), NC–ASiO and C–ASiO electrodes.
Results and discussion
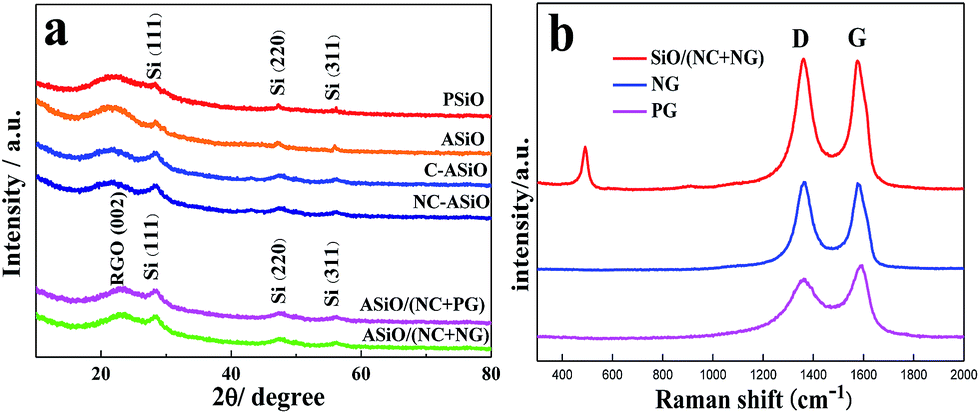
The XRD patterns of PSiO, ASiO, C–ASiO, NC–ASiO, ASiO/(NC + PG) and ASiO/(NC + NG) are shown in Fig. 1a. All diffraction peaks seen from the samples exhibit several clear peaks at 28.3°, 47.2°, and 56.1°, which correspond to the (111), (220) and (311) lattice orientations of Si (JCPDS no. 27-1402), respectively. No peaks related to impurities were observed. For the PSiO and ASiO, the high efficient attritor mill process had no significant effect on the XRD peaks, although a slight decrease of the Si peak (28.3°) height and a slight increase of the SiO2 peak (22°) height in the pattern of ASiO were evident. No diffraction peaks corresponding to graphitic carbon were observed in the XRD patterns of C–ASiO and NC–ASiO, meaning that the carbon coating (N-doping) is amorphous. There is no obvious difference between the C–ASiO and NC–ASiO samples from the XRD patterns, indicating that N-doping had not been changed the amorphous structure of C–ASiO. The breadth of the Si peaks in C–ASiO and NC–ASiO samples suggests that Si crystal held in the composite materials would have low crystallinity or be extremely small in size. For the XRD patterns of ASiO/(NC + PG) and ASiO/(NC + NG), the similar broad reflections due to 002 planes of graphite suggest the presence of largely exfoliated graphene layers. It is also can be found that no obvious change corresponding to the incorporation of N atoms within the NG sheets were found owing to the low N-doping content.26
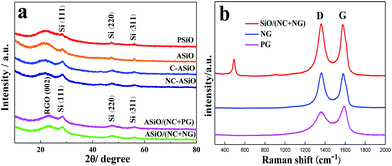 |
| | Fig. 1 (a) XRD patterns of PSiO, ASiO, C–ASiO, NC–ASiO, ASiO/(NC + PG) and ASiO/(NC + NG); (b) Raman spectra of PG, NG and ASiO/(NC + NG). | |
Raman spectroscopy is a powerful non-destructive technique to probe structural and electronic characteristics of graphite materials,32 particularly to determine the defects, the ordered and disordered structures. Fig. 1b presents the Raman spectrum of ASiO/(NC + NG), in which the Raman spectra of PG and NG were also provided for comparison. The Raman spectrum of the ASiO/(NC + NG) displays a broad peak at around 491 cm−1, which is due to the SiO nanoparticles.33 In accordance with the previous literature results,67 this results shows that Si nanostructure existed in ASiO samples distributed homogeneously. Another two prominent peaks at around 1362 cm−1 and 1576 cm−1, which are identified respectively as the D band and G band of NG.31 The intensity of the D band is strongly associated with disordered carbon, edge defects and other defects (sp3 bonded carbon, dangling bonds, vacancies, and topological defects), while the G band corresponds to the zone center E2g mode related to phonon vibrations in sp2 carbon materials.34 The Raman spectrum of PG exhibit two remarkable peaks at around 1362 and 1588 cm−1 corresponding to the well-defined D band and G band, respectively. In comparison with PG, the G band of NG exhibits an up-shift of 8 cm−1, which is similar to previously reported doped graphitic materials.35–37 Frequently, the intensity ratio of D band to G band (ID/IG) is used to estimate the disorder of graphene,35,38 which consists with the sp3/sp2 carbon ratio.39 The ID/IG of the band intensities for NG (1.09) has increased compared to the PG (0.78), suggesting that pronounced increase in disorder has occurred due to N atoms are incorporated into the graphene network.24 Therefore, the relatively increased ID/IG and upshift of the G band observed for the NG provide evidence for the successful N-doping in NG. Table S1† lists the elemental composition of different samples. The result of elemental content analysis confirmed that the ASiO content of ASiO/(NC + NG), ASiO/(NC + PG), NC–ASiO and C–ASiO were 60.39 wt%, 60.41 wt%, 86.29 wt% and 86.9 wt%, respectively (calculated on the basis of stoichiometric proportion of SiO). In addition, from the results of the four-probe conductivity measurement, the electric conductivities of C–ASiO and NC–ASiO are 8.53 × 10−4 S cm−1 and 1.79 × 10−3 S cm−1, respectively, suggesting that the simple N-doping in the surface carbon coating layers enhanced the conductivity of as prepared ASiO anode more significantly as compared with undoped carbon coating mode.
In order to determine the chemical composition of ASiO/(NC + NG) composite, XPS measurements were carried out. As shown in Fig. S1,† XPS survey scan of NC–ASiO clearly shows the presence of C 1s, Si 2p, N 1s and O 1s with no trace of impurities. Combined with the results of TEM image (Fig. 3a), HRTEM image (Fig. 3b) and XPS survey spectrum of NC–ASiO (Fig. S1†), it is indicated that ASiO nanoparticles were successfully coated with N-doped carbon surface layers. As shown in Fig. 2a, the XPS survey spectrum demonstrates four dominant peaks centered at 103.6, 285.2, 399.6 and 531.6 eV, corresponding to C (C 1s), Si (Si 2p), N (N 1s) and O (O 1s) and the atomic ratio of the elements is summarized in the insert of Fig. 2a. Fig. 2b shows the high-resolution XPS spectrum of Si 2p, which fits into five peaks, corresponding to five different Si oxidation states (Si0, Si+, Si2+, Si3+ and Si4+), on the basis of binding energy values previously reported.40,41 For the XPS result of Si 2p, Si2+ and Si3+ were the main peaks. The average valence state of Si on the ASiO/NC + NG composite is 2.09 and the calculated atomic ratio of Si to O element is about 1 to 1, approaching the theoretical value of SiO. Energy dispersive X-ray analysis (EDAX) (Fig. S2†) verifies the existence of element Si and O with approximately 1:1 ratio, indicating the product is stoichiometric SiO. High-resolution C 1s spectrum of ASiO/NC + NG composite is shown in Fig. 3c. The C 1s spectrum has four components centered at 284.6, 286.3, 287.8 and 288.8 eV. The main peak at 284.6 eV (labeled as C1) is corresponded to the graphite-like sp2 C, which indicates that most of the C atoms in the ASiO/(NC + NG) composite are arranged in a conjugated honeycomb lattice. Small peaks at 286.3 eV (labeled as C2) and 287.8 eV (labeled as C3) are clearly visualized in addition to the main sp2 C peak, reflecting two different bonds, corresponding to the C–OH (C–O–C) and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, respectively. The peaks located at higher energy in the C1s spectrum suggests the nitrogen doping occurs in the RGO.42 The C 1s peak at 288.8 eV is attributed to O–CO bond.42,43 This peak is ascribed to the physisorbed oxygen on the RGO. As shown in Fig. 2d, the high-resolution N 1s spectrum of ASiO/(NC + NG) composite can be splited into four individual peaks that are assigned to pyridinic N (398.5 eV) (labeled as N1), pyrrolic N (400.1 eV) (labeled as N2), graphitic N (401.3 eV) (labeled as N3) and N-oxides of pyridinic N (402.5 eV) (labeled as N4).45–48 The pyridinic N that contributes to the π system with one p electron corresponds to a tetrahedral nitrogen phase bonded to a sp3-hybridized carbon atom (N-sp3 C).44 The pyrrolic N atoms with two p electrons in the π system corresponds to a trigonal nitrogen phase bonded to a sp2-coordinated carbon atom (N-sp2 C).44 For graphitic N, in which nitrogen atoms are incorporated into the graphene layers and replace carbon atoms within a graphene plane; N-oxides of pyridinic-N are bonded to two carbon atoms and one oxygen atom. A doping level of 3.2 at% N content in the ASiO/(NC + NG) composite was obtained and the N binding configuration includes 44.44% pyridinic N, 28.57% pyrrolic N, 17.46% graphitic N and 9.52% N-oxides of pyridinic-N.
O, respectively. The peaks located at higher energy in the C1s spectrum suggests the nitrogen doping occurs in the RGO.42 The C 1s peak at 288.8 eV is attributed to O–CO bond.42,43 This peak is ascribed to the physisorbed oxygen on the RGO. As shown in Fig. 2d, the high-resolution N 1s spectrum of ASiO/(NC + NG) composite can be splited into four individual peaks that are assigned to pyridinic N (398.5 eV) (labeled as N1), pyrrolic N (400.1 eV) (labeled as N2), graphitic N (401.3 eV) (labeled as N3) and N-oxides of pyridinic N (402.5 eV) (labeled as N4).45–48 The pyridinic N that contributes to the π system with one p electron corresponds to a tetrahedral nitrogen phase bonded to a sp3-hybridized carbon atom (N-sp3 C).44 The pyrrolic N atoms with two p electrons in the π system corresponds to a trigonal nitrogen phase bonded to a sp2-coordinated carbon atom (N-sp2 C).44 For graphitic N, in which nitrogen atoms are incorporated into the graphene layers and replace carbon atoms within a graphene plane; N-oxides of pyridinic-N are bonded to two carbon atoms and one oxygen atom. A doping level of 3.2 at% N content in the ASiO/(NC + NG) composite was obtained and the N binding configuration includes 44.44% pyridinic N, 28.57% pyrrolic N, 17.46% graphitic N and 9.52% N-oxides of pyridinic-N.
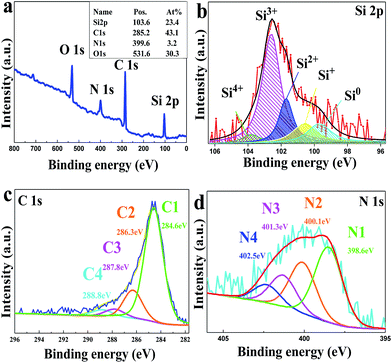 |
| | Fig. 2 XPS patterns of ASiO/(NC + NG) sample: (a) survey and high-resolution of (b) Si 2p, (c) C 1s and (d) N 1s. | |
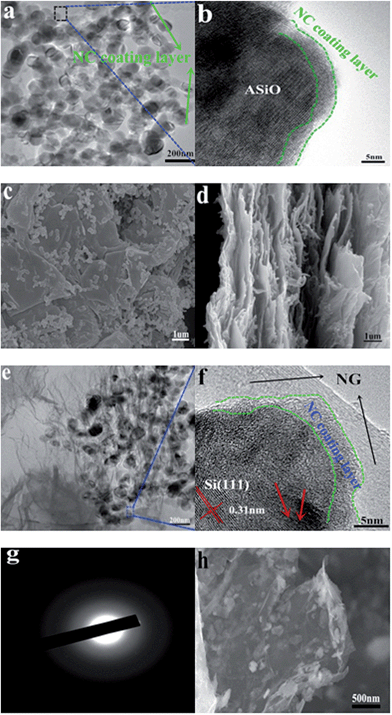 |
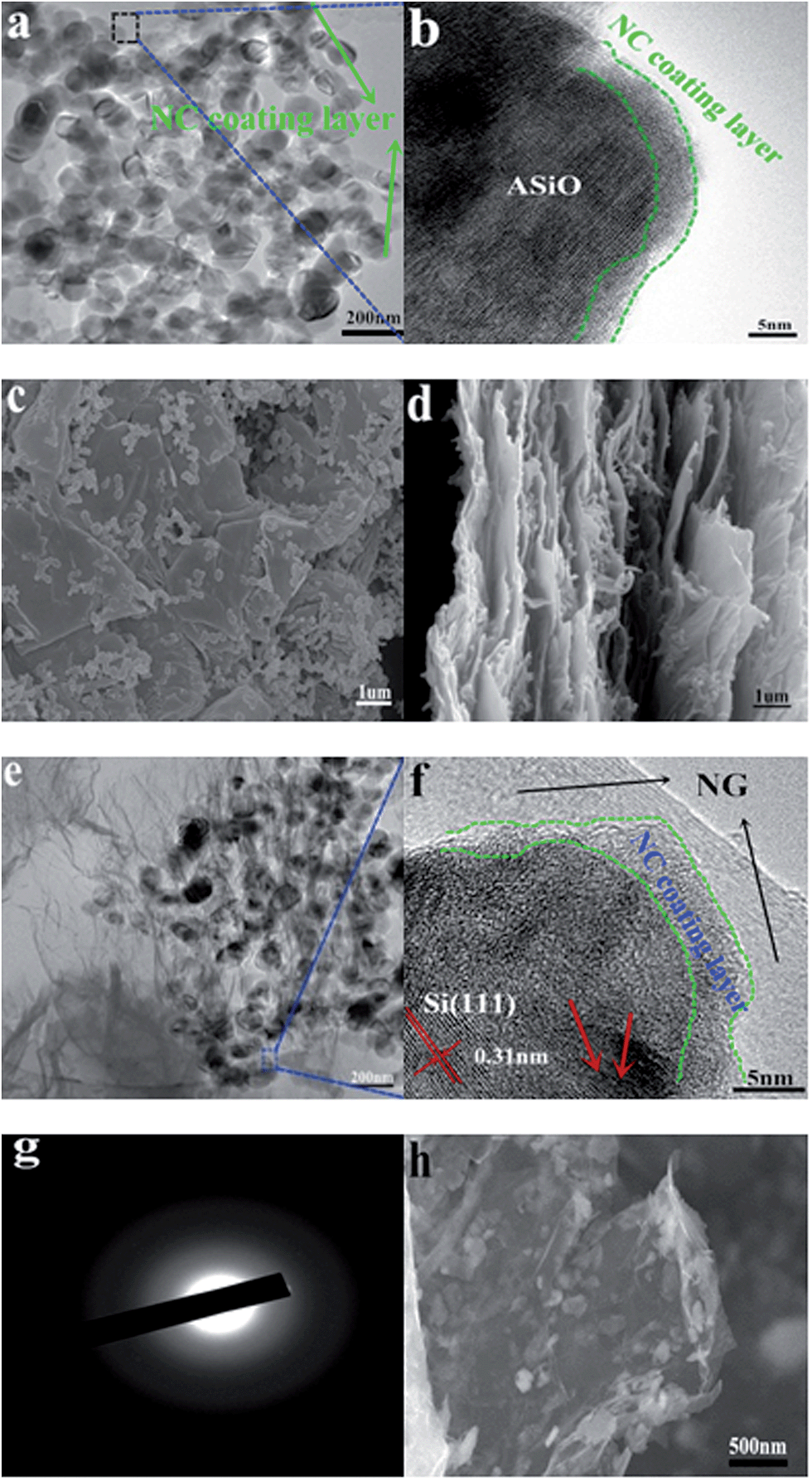
| | Fig. 3 (a) TEM image of NC–ASiO; (b) HRTEM image of NC–ASiO; (c) top-view FESEM image of ASiO/(NC + NG); (d) cross-section view FESEM image of ASiO/(NC + NG); (e) TEM image of ASiO/(NC + NG); (f) HRTEM image of ASiO/(NC + NG); (g) SAED pattern of ASiO/(NC + NG); (h) FESEM image of ASiO/(NC + NG) after 200 cycles under a current density of 100 mA g−1. | |
The size and morphology of the samples were characterized by FESEM, TEM and HRTEM respectively. The FESEM images of as-received SiO powder and ASiO nanoparticles are presented in Fig. S3a and b† in order to compare their morphologies. The initial particle size of 40 μm was reduced to 50–150 nm by high efficient attritor mill process. Samples NC–ASiO nanoparticles and ASiO nanoparticles exhibit a similar morphology (Fig. 3a, S3c and S3b†) consisting of quasi-spherical nanoparticles and have an equable size distribution ranging from 50 to 150 nm. Simultaneously, a random aggregation of primary nanoparticles can also be observed. The NC coating structure was also investigated by HRTEM image, as shown in Fig. 3b (the black rectangle region in Fig. 3a), about 3 nm thickness of amorphous NC coating layer is found on the surface of ASiO particle. FESEM images of as-prepared PG and NG (Fig. S4a and S4b†) show that after N-doping, NG still maintains the two-dimensional ultrathin flexible structure of the PG, but has a surface morphology with more corrugations and scrollings than PG. As shown in the top-view (Fig. 3c) FESEM image of ASiO/NC + NG, NC–ASiO nanoparticles disperse homogeneously on the NG sheets, which are curly and gauze-like. From the cross-section view (Fig. 3d) FESEM image of ASiO/(NC + NG), we could observe the insertion of NC–ASiO nanoparticles between the interlayers of NG sheets and that nanoporous composites with a large number of cavities were formed. The disorderly stacked layer-by-layer structure of NG can also be observed, which demonstrates a feature of 3D nanocomposites. The TEM image of ASiO/(NC + NG) (Fig. 3e) revealed that the NC–ASiO nanoparticles in the range of 50–150 nm were firmly anchored to the surface of NG matrix, wrapped by the NG film or embedded in the NG network. Sheet-like NG with numerous folds at the edges can also be found in this image. Thus, a more effective 3D “sheet-web” mode mixed (electron and ion) conductive network49,50 will be built around the active nanoparticles, which also can enhance the electronic conductivity of active material and decrease the effect of particle-agglomeration phenomenon effectively. In the HRTEM image ASiO/(NC + NG) (Fig. 3f (the blue rectangle region in Fig. 3e)), ASiO nanoparticles in the final products are covered with a thin layer of amorphous NC (3–5 nm) and embedded into the NG network. The clearly visible set of lattice fringes with a period of ∼0.31 nm is the characteristic of (111) lattice planes of Si crystal. The disordered domain (indicated by red arrows) in the nanocomposites should be amorphous SiOx (0 ≤ x ≤ 2) microstructure. The selected-area electron diffraction (SAED) pattern of ASiO/(NC + NG) (Fig. 3g) in this region comprises a well-resolved set of concentric rings with bright spots and confirms the pattern of Si crystal, which is consistent with XRD analysis. Atomic force microscopy (AFM) analysis also further demonstrates that the NC–ASiO nanoparticles are homogeneously dispersed and anchored onto the NG sheets (Fig. S5a and b†). Meanwhile, the cross section analysis of the AFM image is shown in Fig. S5c.† From the line profile of the ASiO/(NC + NG) nanocomposites, the height of the NC–ASiO nanoparticles on the NG sheets is estimated to be about 79.8 nm. This height value is within the range of diameter of NC–ASiO nanoparticles, which is consistent with that in FESEM and TEM images observation. Fig. S6a and b† shows the nitrogen adsorption/desorption isotherm and Barrett–Joyner–Halenda (BJH) curve of the ASiO/(NC + NG) nanocomposites. The isotherm belongs clearly to type IV, and the hysteresis loops indicate that existence of mesoporous in the synthesized nanocomposites (Fig. S6a†). From the adsorption branch of isotherm curves, the specific surface area of 380.896 m2 g−1 is calculated through the multi-point Brunauer–Emmett–Teller (BET) surface area analysis. From the pore size distribution curve (Fig. S6b†), the most probable pore diameter of 2.69 nm with narrow distribution and the average pore diameter of 5.80 nm are calculated using the BJH model. The pore width distributions calculated with the BJH model are concentrated in the range of a typical mesoporous structure. The total pore volumes with pore width from 1.7 to 300 nm are 0.48 cm3 g−1 for ASiO/(NC + NG). The large pore volume could arise primarily from the formation of secondary pores between the NC–ASiO nanoparticles and NG sheets as well as the close stacking of NG sheets distributed between the NC–ASiO nanoparticles. The high surface area and typical mesoporous nanostructure, in association with the co-modified strategy (NC coating layer and NG network), indicate that somewhat enhanced electrochemical performances of anode material in LIBs should be expected.
The electrochemical performance of the samples as anodes for LIBs was tested using CR2016 coin-type half-cells. Fig. 4a delineates the initial discharge and charge curves of various anodes. ASiO/(NC + NG) and ASiO/(NC + PG) electrodes have the similar potential plateaus with NC–ASiO, indicating that PG or NG has no obvious potential plateaus. ASiO/(NC + NG) exhibited a specific discharge capacity of 2465 mA h g−1, which had exceeded the data reported in the literatures.7–12 The extra capacity in initial cycle is probably contributed by the enhanced surface area or topological defects produced during the N-doping process (additional storage of Li+ on the surface or on the edge sites) and the formation of a surface polymeric layer due to the decomposition of the solvent in the electrolyte, which caused the irreversible capacity loss.50 By contrast, for ASiO/(NC + PG), NC–ASiO and C–ASiO, the discharge capacities were 2390.5, 1511 and 1360 mA h g−1 respectively, which were lower than that of ASiO/(NC + NG). Fig. 4b shows CV curves of ASiO/(NC + NG) nanocomposites from the first cycle to the fifth cycle in the potential window of 0.01–3.0 V (vs. Li/Li+) at a scan rate of 0.1 mV s−1. The CV measurement of the ASiO/(NC + NG) electrode showed that lithium could reversibly intercalate and de-intercalate into active materials. As can be seen in the first cycle, two characteristic pairs (cathodic, anodic) of current peaks were observed at the potential of (0.02, 0.78, 1.2 V) and (0.50 V). The results are well consistent with the data reported in the literature.51,52 The reduction peak around 0.78 V and 1.2 V may be ascribed to the formation of SEI film on the surface of active materials, the reduction of SiO2 to Si and the synchronous formation of Li2O and Li4SiO4,9 as described in eqn (1) and (2):
| | |
2SiO2 + 4Li → Li4SiO4 + Si
| (1) |
| | |
SiO2 + 4Li → 2Li2O + Si
| (2) |
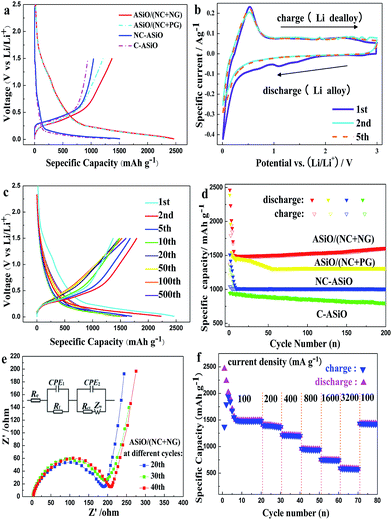 |
| | Fig. 4 (a) Initial charge and discharge curves of ASiO/(NC + NG), ASiO/(NC + PG), NC–ASiO and C–ASiO under a current density of 100 mA g−1. (b) The CV curves of the as-prepared ASiO/(NC + NG) nanocomposites from the first cycle to the fifth cycle measured in the voltage range of 0.01–3.0 V with a scan rate of 0.1 mV s−1; (c) discharge–charge curves of ASiO/(NC + NG) cycled between 0 and 1.5 V under a current density of 100 mA g−1 for the 1st, 2nd, 5th, 10th, 20th, 50th, 100th and 500th cycle; (d) cycling behaviors of ASiO/(NC + NG), ASiO/(NC + PG), NC–ASiO and C–ASiO under a current density of 100 mA g−1; (e) Nyquist plots of the ASiO/(NC + NG) electrode obtained at different cycles; insert shows the equivalent circuit model of the studied system; (f) cycling performances of ASiO/(NC + NG) under different current densities. | |
The lithium insertion potential is quite low, which is very close to 0 V (0.02 V) vs. the Li/Li+ reference electrode. The cathodic peak at about 0.02 V corresponds to the formation of Li–Si alloy phases and the reversible reaction between Li and carbon,53,54 as described in eqn (3) and (4) respectively:
| | |
Si + xLi ↔ LixSi (0 ≤ x ≤ 4.4)
| (3) |
The pronounced peaks at 0.50 V at the anodic sweep could be ascribed to de-alloying of Li–Si alloys, as described in eqn (5):
| | |
LixSi ↔ Si + xLi (0 ≤ x ≤ 4.4)
| (5) |
Fig. 4c displays the discharge–charge profiles of the ASiO/(NC + NG) nanocomposites cycled during the 1st, 2nd, 5th, 10th, 20th, 50th, 100th and 500th cycles between the voltage limits of 0–1.5 V vs. Li/Li+ at a current density of 100 mA g−1. The discharge–charge curves remain similar in shape, though a large irreversible capacity loss was observed in the first cycle, the reversibility of the capacity was significantly improved, with an average columbic efficiency of >98% for up to 500 cycles after the fifth cycle. The cycling performances of the samples under a current density of 100 mA g−1 are presented in Fig. 4d. For ASiO/(NC + NG), from the 1st cycle to the 13th cycle, its reversible specific capacity decreases with cycles and shows the normal characteristics. Interestingly, as the cycling continued from the 13th cycle up to the 200th cycle, its reversible specific capacity increases back and retains at approximately 1603 mA h g−1 in the 200th cycle, which is 4 times higher than the theoretical capacity of graphite. For comparison, the reversible specific capacity of ASiO/(NC + PG), NC–ASiO and C–ASiO in the last recorded cycle are 1313, 1011 and 803 mA h g−1, respectively. Fig. S7† shows excellent cycle stability of ASiO/(NC + NG) nanocomposites in the long-run up to 500 cycles under a current density of 100 mA g−1. After the 200th cycle, the reversible specific capacity continues to increase, and reaches a maximum value (1790 mA h g−1) in the around 446th cycle and then remains constant at around 1790 mA h g−1. The phenomenon of firstly decreasing and afterwards increasing of reversible capacity may be caused by inducing more defect sites and vacancies as Li+ active sites onto surface of graphene sheet.27 This interesting phenomenon can be interpreted by the EIS measurements of the ASiO/(NC + NG) were carried out after 20, 30 and 40 cycles, as shown in Fig. 4e. The Nyquist plots obtained were modeled and analysed with the help of an appropriate electric equivalent circuit. In the equivalent circuit (inset of Fig. 4e), Re represents the total resistance of electrolyte, electrode, and separator. Rf and CPE1 are the resistance and capacitance, respectively, of the SEI film formed on the electrode. Rct and CPE2 represent the charge-transfer resistance and the double layer capacitance, respectively, and Zw is the Warburg impedance related to the diffusion of Li+ into the bulk electrode.55 In Fig. 4e, the high frequency semicircle is corresponding to the resistance Rf and CPE1 of SEI film, the medium frequency semicircle is assigned to the charge-transfer resistance Rct and CPE2 of electrode–electrolyte interface. The sloping line in the lower frequency represents the Warburg impedance, which is associated with Li+ diffusion in the bulk of active materials.56,57 The reasonably good fitting results are presented in Table S2.† On the basis of the equation described below, we could calculate the effective capacitance (C) as follows:58
where
R represents resistance,
Q represents constant phase element, and
η is the empirical constant which can take any arbitrary value between zero and one.
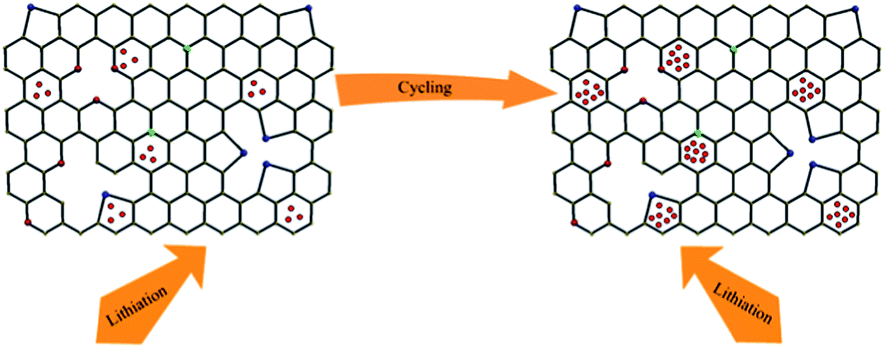
59 As reported in
ref. 63, electrochemical surface area (including surface defects and vacancies) are believed to be responsible for the increased reversible capacity. It was found that the capacitance value (Table S2
†) of the electrode increases during the cycle, which means the electrochemical surface area of the electrode increases. It is indicated that Li
+ intercalation/de-intercalation into ASiO/(NC + NG) nanocomposites upon cycling brings about more defect sites and vacancies (as shown in
Scheme 2).
60 These defect sites and vacancies in ASiO/(NC + NG) nanocomposites provide more Li
+ storage electrochemical active locations, thus delivering the increasing capacity during the subsequent charge–discharge cycles. In addition, the pyridinic N atoms could also result in the improvement of the reversible capacity of the ASiO/(NC + NG) nanocomposites electrode.
24,60 The robust structure of the ASiO/(NC + NG) nanocomposites is confirmed by the FESEM image of the sample after being used in LIBs for 200 cycles (
Fig. 3h). As shown in the image, NC–ASiO nanoparticles remain integrated with NG network after 200 cycles at 100 mA g
−1, showing that the electrode was not damaged by the cycling test.
Fig. 4f shows the rate capabilities of ASiO/(NC + NG) nanocomposites under current densities of 100 mA g
−1, 200 mA g
−1, 400 mA g
−1, 800 mA g
−1, 1600 mA g
−1 and 3200 mA g
−1. Obviously, the ASiO/(NC + NG) nanocomposites also demonstrated good rate performance, even under the very high current density of 3200 mA g
−1, it retained a high reversible specific capacity of about 580 mA h g
−1. The observed rate capability is among one of the highest values so far reported for SiO-based electrodes.
8–13,52,62,63 After the current density returned to 100 mA g
−1, the ASiO/(NC + NG) nanocomposites delivered a reversible specific capacity of about 1425 mA h g
−1, showing that the electrode was not damaged by the high rate test. The results of high rate electrochemical studies prove that the ASiO/(NC + NG) electrode could be used as an excellent high rate electrode.
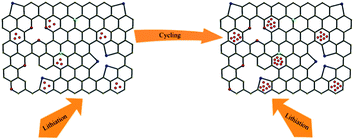 |
| | Scheme 2 A schematic model of Li storage in defects and vacancies according to ref. 60. | |
The above electrochemical data demonstrated the improved electrochemical performance of ASiO/(NC + NG), which can be attributed to four main factors. Firstly, nanosized composites and mesoporous structure are a prerequisite and the reasons are as follows: (i) the higher interfacial area with the electrolyte and hence a higher flux across the interface; (ii) the shorter lengths of the ion and electron transport; (iii) the strain associated with Li+ intercalation/de-intercalation being better accommodated by smaller nanocomposites due to the larger pore volumes surrounding them; (iv) the possibility of forming a 3D mixed conducting network in which mesopores structure allow both Li+ and electron to migrate rapidly. Secondly, the presence of NC coating layer could improve the interfacial electric conductivity, resulting from a remarkable electron transfer from the SiO surface to the coating layers, yielding excellent electrochemical properties for energy storage applications. Thirdly, the NG sheet preserve the advantageous characteristics of graphene, such as an ultrathin framework, open porous structure, high surface area, mechanical flexibility, and chemical stability.64–66 Moreover, the defect sites and vacancies produced during the N-doping process and oxygen-containing groups may enable the NG to be favorable for Li+ storage and consequently improved the electrochemical properties of the ASiO/(NC + NG) nanocomposites.24,61 Last but not least, the double protection strategy (co-modification of NC coating layer and NG network) could effectively constructed a 3D conducting network through a 3D “sheet-web” mode and the preeminent electrochemical activity of ASiO/(NC + NG) in this study can be attributed to the synergistic effect of NC coating layer and NG network as follows: (i) the NC coating layer functions as a stabilizer to immobilize ASiO nanoparticles, a buffer to play an important role in minimizing volume changes and direct contact between ASiO nanoparticles and the electrolyte, facilitating the formation of a stable SEI film. Therefore mechanical stability and electrical contact were increased for ASiO/(NC + NG) nanocomposites; (ii) due to its distinctive properties, NG network which serve as the elastic and electronical conductive substrate, afford good dispersion of NC–ASiO nanoparticles and maintain a high electrical conductivity of the whole material, thus realizing dramatically improved reversible specific capacity, cycling performance and rate capability as an anode material for LIBs.
Conclusions
In summary, a 3D ASiO/(NC + NG) nanocomposites was synthesized successfully as an anode for LIBs by the ASiO nanoparticles further co-modified with NC coating layer and NG network. The exceptional high reversible specific capacity, ultra long cyclic life and superior high-rate capability of as synthesized material can be related to reduced active particle size, the enhancement of conductivity by NC coating layer, the extension of the defect sites and vacancies of the NG network and the synergistic effect of NC coating layer and NG network, which helps to keep the entire structure stable during charge/discharge process. The ASiO/(NC + NG) nanocomposites could be further explored for high-capacity and environment-friendly anode material for LIBs applications. In addition, the economical and simple HEAM technique (benefiting from its high yield and high efficiency), special ionic liquid as a new type of precursor for NC coating technique (benefiting from ionic liquid's almost ideal precursor properties) and the thermal annealing technique for synthesis of NG (benefiting from its properties of facile and catalyst-free) confirmed the technological feasibility of preparation process. This preparation strategy is of potential interest to other anode materials with large volume variations and low electrical conductivities in the battery area.
Acknowledgements
We appreciate the support of National Natural Science Foundation of China (no. 50974045).
Notes and references
- J. Maier, Nat. Mater., 2005, 4, 805 CrossRef CAS.
- A. S. Aricò, P. Bruce, B. Scrosati, J.-M. Tarascon and W. van Schalkwijk, Nat. Mater., 2005, 4, 366 CrossRef PubMed.
- M. Armand and J.-M. Tarascon, Nature, 2008, 451, 652 CrossRef CAS PubMed.
- J.-H. Kim, C.-M. Park, H. Kim, Y.-J. Kim and H.-J. Sohn, J. Electroanal. Chem., 2011, 661, 245 CrossRef CAS PubMed.
- S. D. Beattie, D. Larcher, M. Morcrette, B. Simon and J. M. Tarascon, J. Electrochem. Soc., 2008, 155, A158 CrossRef CAS PubMed.
- L. Y. Beaulieu, K. W. Eberman, R. L. Turner, L. J. Krause and J. R. Dahn, Electrochem. Solid-State Lett., 2001, 4, A137 CrossRef CAS PubMed.
- Y.-J. Chao, X. Yuan and Z.-F. Ma, Electrochim. Acta, 2008, 53, 3468 CrossRef CAS PubMed.
- C.-H. Doh, C.-W. Park, H.-M. Shin, D.-H. Kim, Y.-D. Chung, S.-I. Moon, B.-S. Jin, H.-S. Kim and A. Veluchamy, J. Power Sources, 2008, 179, 367 CrossRef CAS PubMed.
- Q. Si, K. Hanai, T. Ichikawa, M. B. Phillipps, A. Hirano, N. Imanishi, O. Yamamoto and Y. Takeda, J. Power Sources, 2011, 196, 9774 CrossRef CAS PubMed.
- M. Miyachi, H. Yamamoto and H. Kawai, J. Electrochem. Soc., 2007, 154(4), A376 CrossRef CAS PubMed.
- T. Tabuchi, H. Yasuda and M. Yamachi, J. Power Sources, 2005, 146, 507 CrossRef CAS PubMed.
- D. Jin Lee, M.-H. Ryou, J.-N. Lee, B. Gon Kim, Y. Min Lee, H.-W. Kim, B.-S. Kong, J.-K. Park and J. W. Choi, Electrochem. Commun., 2013, 34, 98 CrossRef PubMed.
- S. Komaba, K. Shimomura, N. Yabuuchi, T. Ozeki, H. Yui and K. Konno, J. Phys. Chem. C, 2011, 115, 13487 CAS.
- A. Guerfi, P. Charest, M. Dontigny, J. Trottier, M. Lagacé, P. Hovington, A. Vijh and K. Zaghib, J. Power Sources, 2011, 196, 5667 CrossRef CAS PubMed.
- J. Wang, H. Zhao, J. He, C. Wang and J. Wang, J. Power Sources, 2011, 196, 4811 CrossRef CAS PubMed.
- C. Guo, D. Wang, Q. Wang, B. Wang and T. Liu, Int. J. Electrochem. Sci., 2012, 7, 8745 CAS.
- Z. Ding, L. Zhao, L. Suo, Y. Jiao, S. Meng, Y.-S. Hu, Z. Wang and L. Chen, Phys. Chem. Chem. Phys., 2011, 13, 15127 RSC.
- Y. Ma, C. Zhang, G. Ji and J. Y. Lee, J. Mater. Chem., 2012, 22, 7845 RSC.
- L. Zhao, Y.-S. Hu, H. Li, Z. Wang and L. Chen, Adv. Mater., 2011, 23, 1385 CrossRef CAS PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666 CrossRef CAS PubMed.
- A. K. Geim and K.
S. Novoselov, The Rise of Graphene, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- A. K. Geim, Science, 2009, 324, 1530 CrossRef CAS PubMed.
- H. Wang, C. Zhang, Z. Liu, L. Wang, P. Han, H. Xu, K. Zhang, S. Dong, J. Yao and G. Cui, J. Mater. Chem., 2011, 21, 5430 RSC.
- A. L. M. Reddy, A. Srivastava, S. R. Gowda, H. Gullapalli, M. Dubey and P. M. Ajayan, ACS Nano, 2010, 4(11), 6337 CrossRef CAS PubMed.
- Z.-S. Wu, W. Ren, L. Xu, F. Li and H.-M. Cheng, ACS Nano, 2011, 5(7), 5463 CrossRef CAS PubMed.
- Y. Shao, S. Zhang, M. H. Engelhard, G. Li, G. Shao, Y. Wang, J. Liu, I. A. Aksay and Y. Lin, J. Mater. Chem., 2010, 20, 7491 RSC.
- X. Li, D. Geng, Y. Zhang, X. Meng, R. Li and X. Sun, Electrochem. Commun., 2011, 13, 822 CrossRef CAS PubMed.
- C. Guo, D. Wang, T. Liu, J. Zhu and X. Lang, J. Mater. Chem. A, 2014, 2, 3521 CAS.
- L. Wang and D.-L. Wang, Electrochim. Acta, 2011, 56, 5010 CrossRef CAS PubMed.
- B. Wang, D. Wang, Q. Wang, T. Liu, C. Guo and X. S. Zhao, J. Mater. Chem. A, 2013, 1, 135 CAS.
- Z.-H. Sheng, L. Shao, J.-J. Chen, W.-J. Bao, F.-B. Wang and X.-H. Xia, ACS Nano, 2011, 5(6), 4350 CrossRef CAS PubMed.
- A. Das, S. Pisana, B. Chakraborty, S. Piscanec, S. K. Saha, U. V. Waghmare, K. S. Novoselov, H. R. Krishnamurthy, A. K. Geim and A. C. Ferrari, Nat. Nanotechnol., 2008, 3, 210 CrossRef CAS PubMed.
- L. Khriachtchev, S. Novikov and O. Kilpelä, J. Appl. Phys., 2000, 87, 7805 CrossRef CAS PubMed.
- K. N. Kudin, B. Ozbas, H. C. Schniepp, R. K. Prud'homme, I. A. Aksay and R. Car, Nano Lett., 2008, 8, 36 CrossRef CAS PubMed.
- B. D. Guo, Q. A. Liu, E. D. Chen, H. W. Zhu, L. A. Fang and J. R. Gong, Nano Lett., 2010, 10, 4975 CrossRef CAS PubMed.
- N. Soin, S. S. Roy, S. Roy, K. S. Hazra, D. S. Misra, T. H. Lim, C. J. Hetherington and J. A. McLaughlin, J. Phys. Chem. C, 2011, 115, 5366 CAS.
- Y. C. Lin, C. Y. Lin and P. W. Chiu, Appl. Phys. Lett., 2010, 96, 133110 CrossRef PubMed.
- Z. S. Wu, W. C. Ren, L. B. Gao, J. P. Zhao, Z. P. Chen, B. L. Liu, D. M. Tang, B. Yu, C. B. Jiang and H. M. Cheng, ACS Nano, 2009, 3, 411 CrossRef CAS PubMed.
- N. Mohanty, A. Nagaraja, J. Armesto and V. Berry, Small, 2010, 6, 226 CrossRef CAS PubMed.
- Y. Yasaka, S. Uenaga, H. Yasutake, M. Takakura, S. Miyazaki and M. Hirose, Mater. Res. Soc. Symp. Proc., 1992, 259, 385 CrossRef.
- G. Hollinger and F. J. Himpsel, Appl. Phys. Lett., 1984, 44, 93 CrossRef CAS PubMed.
- K. Chang, D. Geng, X. Li and J. Yang, Adv. Energy Mater., 2013, 3, 839 CrossRef CAS PubMed.
- J. W. Jang, C. E. Lee, S. C. Lyu, T. J. Lee and C. J. Lee, Appl. Phys. Lett., 2004, 84, 2877 CrossRef CAS PubMed.
- C. Ronning, H. Feldermann, R. Merk, H. Hofsäss, P. Reinke and J. U. Thiele, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 2207 CrossRef CAS.
- D. Marton, K. J. Boyd, A. H. Al-Bayati, S. S. Todorov and J. W. Rabalais, Phys. Rev. Lett., 1994, 73, 118 CrossRef CAS.
- P. H. Matter, L. Zhang and U. S. Ozkan, J. Catal., 2006, 239, 83 CrossRef CAS PubMed.
- S. Kundu, T. C. Nagaiah, W. Xia, Y. Wang, S. V. Dommele, J. H. Bitter, M. Santa, G. Grundmeier, M. Bron, W. Schuhmann and M. Muhler, J. Phys. Chem. C, 2009, 113, 14302 CAS.
- R. Arrigo, M. Hävecker, R. Schlögl and D. S. Su, Chem. Commun., 2008, 4891 RSC.
- C. Su, X. Bu, L. Xu, J. Liu and C. Zhang, Electrochim. Acta, 2012, 64, 190 CrossRef CAS PubMed.
- Y. Wang, Z. S. Feng, J. J. Chen and C. Zhang, Mater. Lett., 2012, 71, 54 CrossRef CAS PubMed.
- G. M. Zhou, D. W. Wang, F. Li, L. L. Zhang, N. Li, Z. S. Wu, L. Wen, G. Q. Lu and H. M. Cheng, Chem. Mater., 2010, 22, 5306 CrossRef CAS.
- T. Zhang, J. Gao, H. P. Zhang, L. C. Yang, Y. P. Wu and H. Q. Wu, Electrochem. Commun., 2007, 9, 886 CrossRef CAS PubMed.
- P. Lian, X. Zhu, H. Xiang, Z. Li, W. Yang and H. Wang, Electrochim. Acta, 2010, 56, 834 CrossRef CAS PubMed.
- X. Wang, X. Zhou, K. Yao, J. Zhang and Z. Liu, Carbon, 2011, 49, 133 CrossRef CAS PubMed.
- P. Guo, H. H. Song and X. H. Chen, Electrochem. Commun., 2009, 11, 1320 CrossRef CAS PubMed.
- S. Y. Chung, J. T. Bloking and Y. M. Chiang, Nat. Mater., 2002, 1, 123 CrossRef CAS PubMed.
- Y. Shi, S. L. Chou, J. Z. Wang, D. Wexler, H. J. Li, H. K. Liu and Y. Wu, J. Mater. Chem., 2012, 22, 16465 RSC.
- E. Chinarro, J. R. Jurado, F. M. Figueiredo and J. R. Frade, Solid State Ionics, 2003, 160, 161 CrossRef CAS.
- T. L. Spencer, A. Ramzy, G. R. Goward and V. Thangadurai, Chem. Mater., 2011, 23, 3105 CrossRef CAS.
- N. A. Kaskhedikar and J. Maier, Adv. Mater., 2009, 21, 2664 CrossRef CAS PubMed.
- Y. F. Li, Z. Zhou and L. B. Wang, J. Chem. Phys., 2008, 129, 104703 CrossRef CAS PubMed.
- X. Feng, J. Yang, X. Yu, J. Wang and Y. Nuli, J. Solid State Electrochem., 2013, 17, 2461 CrossRef CAS.
- B.-C. Yu, Y. Hwa, C.-M. Park and H.-J. Sohn, J. Mater. Chem. A, 2013, 1, 4820 CAS.
- E. Yoo, J. Kim, E. Hosono, H. Zhou, T. Kudo and I. Honma, Nano Lett., 2008, 8, 2277 CrossRef CAS PubMed.
- P. Lian, X. Zhu, S. Liang, Z. Li, W. Yang and H. Wang, Electrochim. Acta, 2010, 5, 3909 CrossRef PubMed.
- G. G. Wallace, C. Y. Wang, D. Li and C. O. Too, Chem. Mater., 2009, 21, 2604 CrossRef.
- D. Nesheva, C. Raptis, A. Perakis, I. Bineva and Z. Aneva, J. Appl. Phys., 2002, 92, 4678 CrossRef CAS PubMed.
- J. Yang, Y. Takeda, N. Imanishi, C. Capiglia, J. Y. Xie and O. Yamamoto, Solid State Ionics, 2002, 125, 152 Search PubMed.
- M. Miyachi, H. Yamamoto and H. Kawai, J. Electrochem. Soc., 2007, 154, A376 CrossRef CAS PubMed.
- T. Kim, S. Park and S. M. Oh, J. Electrochem. Soc., 2007, 154, A1112 CrossRef CAS PubMed.
- Y. Nagao, H. Sakaguchi, H. Honda, T. Fukunaga and T. Esaka, J. Electrochem. Soc., 2004, 151, A1572 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra06678b |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.