
Sticky superhydrophobic hard nanofibers from soft matter
Thierry Darmanina,
Claudio Mortiera,
Julian Eastoeb,
Masanobu Sagisakac and
Frederic Guittard*a
aUniv. Nice Sophia Antipolis, LPMC, CNRS, UMR 7336, Parc Valrose, 06100 Nice, France. E-mail: Frederic.GUITTARD@unice.fr; Fax: +33-4-92-07-61-56; Tel: +33-4-92-07-61-59
bSchool of Chemistry, University of Bristol, Bristol, BS8 1TS, UK
cDepartment of Frontier Materials Chemistry, Graduate School of Science and Technology, Hirosaki University, Japan
First published on 30th July 2014
Abstract
Here, we show the possibility to change superhydrophobic properties from soft to hard polymer nanofibers by the control of molecular structure and branching. In fact, we report the synthesis of original monomers derived from 3,4-propylenedioxythiophene (ProDOT) and bearing two branched alkyl chains and their electrodeposition by cyclic voltammetry. We point out that the hydrocarbon moiety (cheaper, readily available, non-toxic) can be an alternative to long fluorocarbon chains (expensive, requiring extensive synthesis, bioaccumulable) to achieve anti-wetting properties. Moreover, we show that the change in the size of branched chains can affect the surface morphology, from soft to hard nanofibers with a high increase in water adhesion due to a lower intrinsic hydrophobicity (sticky superhydrophobicity or parahydrophobicity). We demonstrate the possibility to produce them from soft matter, i.e. polymers. In the case of the hard nanofibers, cross-section images reveal that these fibers are vertically aligned to the substrate. Moreover, we show that the height and the diameter of the hard nanofibers, as well as the distance between the fibers can be controlled by the number of deposition scans. Such materials could be used for biomedical applications, for example.
1 Introduction
Tuning the characteristics and hydrophilic/hydrophobic properties of nanofibers grown on surfaces is crucial for various applications such as cell,1,2 protein3,4 or bacteria adhesion,5 tissue engineering,6 membranes,7 or encapsulation.8 In the case of generation of superhydrophobicity, the wetting properties of surfaces9,10 containing nanofibers highly depend on their intrinsic hydrophobicity, their characteristics (length, diameter), their orientation to the surface (horizontally, vertically) as well as the distance between them.11 Hence, it is extremely important to find a way to control these characteristics.Conducting polymers can be used to produce nanofibers.12 While it was shown the possibility to produce nanofibers in solution,13–17 it was also possible to induce the growth of nanofibers directly on substrates by self-assembly18–23 or electrodeposition.24,25 The electrodeposition process allows control over surface morphology simply by adjusting electrochemical parameters or by designing monomers. Various hydrophobic substituents can be grafted on the monomers to modify the intrinsic polymer hydrophobicity. For the growth of nanofibers, polyaniline,26,27 polypyrrole,28–30 poly(3,4-ethylenedioxythiophene) (PEDOT)31–35 or poly(3,4-propylenedioxythiophene) (PProDOT)36–38 derivatives have been described in the literature. One of the advantages of using ProDOT derivatives is the various possible positions for controlled substitution by hydrophobic substituents. Moreover, when ProDOT is substituted by two similar substituents in the 3-position, the possible configurations (head-to-head, tail-to-tail and head-to-tail) induced during the polymerization lead to the same polymer configuration, because the monomer is symmetric and does not contain an asymmetric carbon. As a consequence, the electrodeposited polymer is more ordered, which is also an advantage for the homogeneity of nanofiber growth. Another advantage is that the surface morphology is highly dependent on the nature of the substituent, and especially its intrinsic hydrophobicity. Hence, both fluorinated and alkyl chains have been used in the literature.28–38 In the case of long fluorinated chains, our group is trying to replace them due to their bioaccumulative potential reported in animals and humans.39,40 The specific interactions between long fluorinated chains, their high insolubility and chemical resistance mean that they cannot be readily metabolized. Moreover, it is necessary that the hydrophobicity of the substituent is not too high in order to preserve the nanofiber morphology.36a To decrease the hydrophobicity of fluorinated or hydrocarbon chain, a way is to introduce branching, which decreases the interchain interactions. In the case of alkyl chains, linear alkyl chains were reported35,36 but not branched analogues.
The use of branched alkyl chains for lowering surface energy has been clearly demonstrated for surfactants in solution41,42 but not yet for treated material surfaces. Moreover, the branching of alkyl chains can also modify the properties of conducting polymers43,44 and affect the surface morphology.
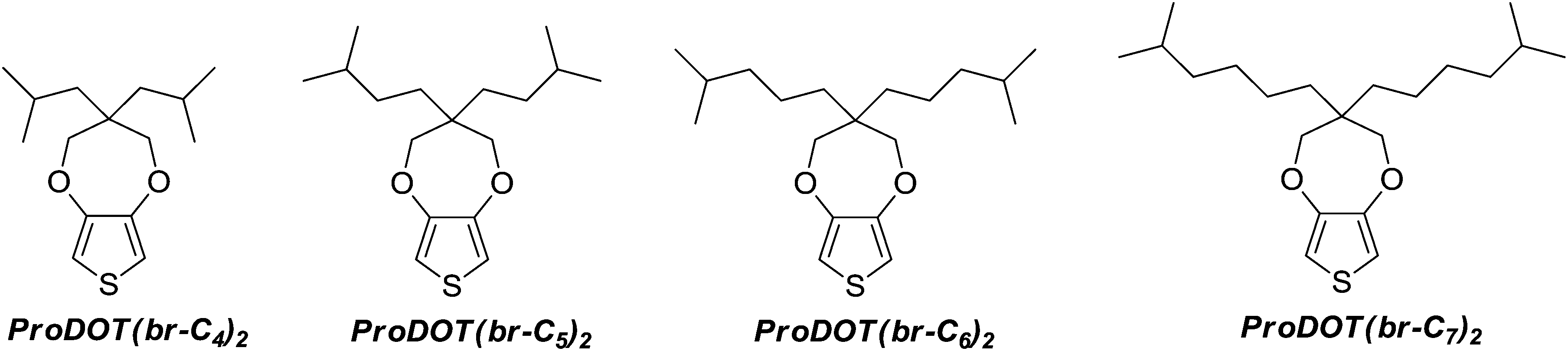
Here, the first study of electropolymerizible monomers with branched alkyl chains is reported. Four original ProDOT derivatives containing two branched alkyl chains have been synthesized, as represented in Schemes 1 and 2, and the surface properties (wettability and morphology) of the corresponding electrodeposited polymers have been studied. It is shown how the size of the branched alkyl chains can be used to obtain soft and hard nanofibers.
 | ||
| Scheme 1 Monomers synthesized in this work. | ||
 | ||
| Scheme 2 Synthetic route to monomers. | ||
2 Experimental
2.1 Monomer synthesis and characterization
The monomers were synthesized in four steps from diethyl malonate (1) and following the synthesis way represented in Scheme 2.2,2-Diisobutyl-1,3-propanediol and 2,2-diisopentyl-1,3-propanediol were purchased from TCI Europe N.V. The other diols (3a and 3b) were obtained by nucleophilic substitution of two hydrogen of diethyl malonate by the corresponding bromoalkane, and the reduction of the two ester groups with lithium aluminum hydride (AlLiH4).45 The general procedure is given below.
Diethyl 2,2-bis(4-methylpentyl)malonate (2a). Yield 25%; colourless liquid; δH(200 MHz, CDCl3, ppm): 4.16 (4H, q, J 7.1), 1.83 (4H, m), 1.54 (2H, quint, 6.5), 1.23 (6H, t, J 7.1), 1.16 (8H, m), 0.84 (12H, d, J 6.5).
Diethyl 2,2-bis(5-methylhexyl)malonate (2b). Yield 20%; colourless liquid; δH(200 MHz, CDCl3, ppm): 4.16 (4H, q, J 7.1), 1.85 (4H, m), 1.50 (2H, quint, J 6.6), 1.23 (6H, t, J 7.1), 1.16 (12H, m), 0.85 (12H, d, J 6.6).
2,2-Bis(4-methylpentyl)propanediol (3a). Yield 95%; colourless liquid; δH(200 MHz, CDCl3, ppm): 3.58 (4H, s), 2.05 (2H, s), 1.55 (2H, m), 1.21 (12H, m), 0.87 (12H, d, J 6.6).
2,2-Bis(5-methylhexyl)propanediol (3b). Yield 95%; colourless liquid; δH(200 MHz, CDCl3, ppm): 3.57 (s, 4H), 2.00 (s, 2H), 1.51 (m, 2H), 1.22 (m, 16H), 0.85 (d, 3JHH = 6.6 Hz, 12H).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).
:1).
3,3-Diisobutyl-3,4-dihydro-2H-thieno[3,4-b][1,4]dioxepine (ProDOT(br-C4)2). Yield 90%; crystalline solid; m.p. 28.7 °C; δH(200 MHz, CDCl3, ppm): 6.42 (2H, s), 3.93 (4H, s), 1.77 (2H, m), 1.42 (4H, d, J 5.6), 0.96 (12H, d, J 6.6); δC(200 MHz, CDCl3, ppm): 149.67, 104.39, 78.27, 45.45, 42.26, 25.38, 23.39; FTIR (KBr): νmax/cm−1 3114, 2959, 2928, 2870, 1486, 1455, 1377, 1191, 1024; MS (70 eV): m/z 268 (M+, 100), 155 (C7H7O2S+, 4), 141 (C6H5O2S+, 55), 127 (C4H7OS+, 25), 116 (C4H4O2S+, 38).
3,3-Diisopentyl-3,4-dihydro-2H-thieno[3,4-b][1,4]dioxepine (ProDOT(br-C5)2). Yield 51%; crystalline solid; m.p. 30.6 °C; δH(200 MHz, CDCl3, ppm): 6.42 (2H, s), 3.84 (4H, s), 1.49 (2H, m), 1.38 (4H, m), 1.12 (4H, m), 1.03 (12H, d, J 6.6); δC(200 MHz, CDCl3, ppm): 149.70, 104.61, 77.58, 43.46, 31.66, 29.29, 28.74, 22.60; FTIR (KBr): νmax/cm−1 3111, 2955, 2928, 2866, 1484, 1455, 1375, 1189, 1020; MS (70 eV): m/z 296 (M+, 100), 155 (C7H7O2S+, 4), 141 (C6H5O2S+, 15), 127 (C4H7OS+, 10), 116 (C4H4O2S+, 30).
3,3-Bis(4-methylpentyl)-3,4-dihydro-2H-thieno[3,4-b][1,4]dioxepine (ProDOT(br-C6)2). Yield 44%; colourless liquid; δH(200 MHz, CDCl3, ppm): 6.42 (2H, s), 3.85 (4H, s), 1.55 (2H, m), 1.25 (12H, m), 0.87 (12H, d, J 6.6); δC(200 MHz, CDCl3, ppm): 149.71, 104.62, 77.49, 43.80, 39.73, 32.01, 27.75, 22.59, 20.49; FTIR (KBr): νmax/cm−1 3115, 2955, 2870, 1486, 1375, 1189, 1024; MS (70 eV): m/z 324 (M+, 100), 155 (C7H7O2S+, 4), 141 (C6H5O2S+, 11), 127 (C4H7OS+, 27), 116 (C4H4O2S+, 52).
3,3-Bis(5-methylhexyl)-3,4-dihydro-2H-thieno[3,4-b][1,4]dioxepine (ProDOT(br-C7)2). Yield 40%; Colourless liquid; δH(200 MHz, CDCl3, ppm): 6.42 (2H, s), 3.84 (4H, s), 1.52 (12H, m), 1.25 (16H, m), 0.86 (12H, d, J 6.6); δC(200 MHz, CDCl3, ppm): 149.70, 104.64, 77.53, 43.72, 38.85, 31.87, 28.22, 27.96, 23.06, 22.61; FTIR (KBr): νmax/cm−1 3114, 2955, 2866, 1485, 1377, 1187, 1024; MS (70 eV): m/z 352 (M+, 100), 155 (C7H7O2S+, 4), 141 (C6H5O2S+, 17), 127 (C4H7OS+, 22), 116 (C4H4O2S+, 49).
2.2 Electropolymerization
Gold plates (with pre-deposited 20 nm chromium and 150 nm gold on silicon wafers) were purchased from Neyco. The electropolymerization process used a three-electrode system: a gold plate as working electrode, a glassy carbon rod as counter-electrode and a saturated calomel reference electrode (SCE). The three-electrode system was connected to an Autolab potentiostat (Metrohm). The electrodes were inserted in a glass cell, in which 10 mL of anhydrous acetonitrile containing 0.1 M of tetrabutylammonium perchlorate (Bu4NClO4) and 0.01 M of monomer were added. The solution was then degassed with argon. The monomer oxidation potential was determined by cyclic voltammetry (Eox = 1.52–1.59 V vs. SCE following the monomer used). Then, cyclic voltammetry was used as the deposition method because it induced the formation of highly homogenous films with high adhesion. With this method, one, three and five scans were performed and with a scan rate of 20 mV s−1. An example of cyclic voltammogram is given in Fig. 1. | ||
| Fig. 1 Cyclic voltammogram of ProDOT(br-C6)2 (0.01 M) recorded in anhydrous acetonitrile containing 0.1 M of Bu4NClO4; Scan rate: 20 mV s−1; Number of scans: 5. | ||
2.3 Surface characterization
The surface morphology was investigated by scanning electron microscopy (SEM) using a 6700F microscope (JEOL). The hydrophobicity of the polymer films was investigated by water contact angle measurements using a DSA30 goniometer (Krüss). The static contact angles were obtained with the sessile drop method, whereas the dynamic contact angles were obtained with the tilted-drop method. Using this last method, the advanced and receding contact angles, and as a consequence the hysteresis (H), were determined after surface inclination and just before the water droplets roll off the surface. The maximum surface inclination is called sliding angle (α).3 Results and discussion
3.1 Surface hydrophobicity
The polymers were electrodeposited by cyclic voltammetry because it induced the formation of highly homogenous films with high adhesion. With this method, one, three and five scans were performed and with a scan rate of 20 mV s−1. In this process, the polymers are obtained in their reduced state (uncharged).The best results were obtained with three deposition scans. For three deposition scans, the apparent contact angles of water (θw) as a function of the number of carbons in the branched alkyl chains (n) are displayed on Fig. 2A. This graph shows that PProDOT(br-C4)2, PProDOT(br-C6)2 and PProDOT(br-C7)2 were superhydrophobic with θw > 150°, but a lower value was obtained for PProDOT(br-C5)2 (θw = 136.8°). Moreover, dynamic contact angle measurements showed low hysteresis (H) and sliding angles (α) for PProDOT(br-C6)2 (H = 5.0° and α = 5.0°) and PProDOT(br-C7)2 (H = 5.7° and α = 4.7°). In an opposite manner, water droplets deposited on PProDOT(br-C4)2 and PProDOT(br-C5)2 remained stuck even after surface inclination of 90° revealing a very high adhesion. To explain these phenomena it was first necessary to explore the surface morphologies.
 | ||
| Fig. 2 Apparent contact angle of water (θw) as a function of the number of carbons in the branched alkyl chains for (A) the structured (3 scans by cyclic voltammetry at 20 mV s−1) and (B) the corresponding smooth films (1 mC cm−2 at imposed potential). | ||
3.2 Surface morphology
Fig. 3 gathers the SEM images of each polymer deposited after three deposition scans. A very unexpected change in the surface morphology was observed from hard polymer nanofibers for PProDOT(br-C4)2 to soft polymer nanofibers for PProDOT(br-C6)2 and PProDOT(br-C7)2. Here, hard nanofibers can be considered as nanoneedles while soft nanofibers are fibers with highly curved surfaces. The change was observed for PProDOT(br-C5)2, for which the surface was not highly structured explaining the lower value of θw obtained for this polymer. Here, the unexpected result was to obtain hard nanofibers because the polymers are considered as soft materials in comparison to metals and inorganic materials. Indeed, it is extremely difficult to generate vertically aligned polymer nanofibers because of their lateral collapse and coalescence, for example during evaporation, due to capillary forces and low stiffness of the fibers.46–49 | ||
| Fig. 3 SEM images of the polymers at two magnifications (5000× and 25000×): (A and B) PProDOT(br-C4)2, (C and D) PProDOT(br-C5)2, (E and F) PProDOT(br-C6)2, (G and H) PProDOT(br-C7)2; number of scans: 3. The insets show a water droplet on the surfaces. | ||
Such fibers were never observed by electrodeposition even with linear alkyl chains,35,36 which shows an effect of the use of branched alkyl chains to control the hardness of nanofibers. Moreover, the hard nanofibers were vertically aligned to the substrate, which could affect the surface wettability.
Now, it is now possible to explain the results for surface properties in terms of the Wenzel and Cassie–Baxter equations.50,51 Indeed, these two equations can be used to explain superhydrophobic properties but with different adhesions. In the Wenzel state50 (cosθ = rcosθY with r a roughness parameter and θY the Young angle),52 a water droplet is in full contact with the surface. As a consequence, the presence of surface roughness can lead to superhydrophobic properties if the materials are intrinsically hydrophobic (θY > 90°) and reversely. However, the increase in the solid–liquid interface with the surface roughness also induces an increase in H, leading to superhydrophobic properties but with high adhesion. When the surface is rough but also porous, a water droplet can be in the Cassie–Baxter state:51 cosθ = fcosθY + f − 1 with f the solid fraction and (1 − f) the air fraction. A water droplet sits on top of the rough surface but also on air pockets entrapped between the solid and the surface. Here, a surface can be superhydrophobic whatever θY if the surface morphology favors the Cassie–Baxter state. Moreover, in the Cassie–Baxter state the adhesion of water is extremely low due to the presence of air, which induces the presence of a liquid–vapor interface.
Because the Wenzel and Cassie–Baxter equation are depending on θY, it was first necessary to determine these angles by producing smooth surfaces for each polymer. Here, it was possible to obtain smooth films for each polymer by changing the deposition process, and using a deposition at constant potential. Smooth surfaces were obtained using an extremely low deposition charge (Qs = 1 mC cm−2) allowing to cover the substrate without formation of structures. However, to obtain the same polymer, a reduction step by cyclic voltammetry (one back scan from 0.8 V to −0.5 V at 20 mV s−1) was added after the deposition to reduce the polymer. The smoothness of the surfaces was confirmed by determining the surface roughness using an optical profilometry (Table 1 and Fig. 4). Their mean roughness (Ra) was below 10 nm and was quite the same for each polymer. Fig. 2B shows the apparent contact angles obtained on the smooth surfaces. An increase of θY was observed between PProDOT(br-C4)2 and PProDOT(br-C6)2 and a decrease after, which means that for PProDOT(br-C7)2 the surface is saturated by hydrocarbon chains. Moreover, PProDOT(br-C5)2, PProDOT(br-C6)2 and PProDOT(br-C7)2 are intrinsically hydrophobic (θY > 90°), whereas for PProDOT(br-C4)2 θY was close to 90°. Now the results can be explained with the Wenzel and Cassie-Baxter equation. In the case of PProDOT(br-C6)2 and PProDOT(br-C7)2, water droplets deposited on these surfaces were close to the Cassie–Baxter state (low H and α) because of their intrinsic hydrophobicity and because the presence of the nanofibers allows to trap a high amount of air between the droplets and the surface.
| Polymer | Ra [nm] | Rq [nm] |
|---|---|---|
| PProDOT(br-C4)2 | 9.3 | 11.9 |
| PProDOT(br-C5)2 | 9.1 | 11.5 |
| PProDOT(br-C6)2 | 9.5 | 11.9 |
| PProDOT(br-C7)2 | 9.2 | 11.7 |
 | ||
| Fig. 4 3-D image of “smooth” PProDOT(br-C7)2 obtained by optical profilometry. | ||
For PProDOT(br-C5)2, the high hydrophobicity and high adhesion can be explained with the Wenzel equation because the surface is not highly structured and θY is > 90°.
For PProDOT(br-C5)2, θw was above 150° but the adhesion of water is extremely high. However, this state cannot be explained with the Wenzel equation because θY is close to 90° (if θY = 90°, the surface roughness does not affect the surface hydrophobicity). Indeed, if the presence of the hard nanofibers can favor the Cassie–Baxter state, the lower θY increases water penetration inside the rough surfaces. Here, the water droplet was probably in an intermediate state between the Wenzel and the Cassie–Baxter known as an impregnating Cassie–Baxter state (Cassie–Baxter state with high adhesion), as observed on the surface of red roses.53–55 Their surface adhesion is due to the combination of microstructures called micropapillae of 16 μm in diameter and 7 μm in height, which are covered by nanofolds. Similar adhesion properties were also reported for peanut leaves.56 In the literature, this state is often called “sticky superhydrophobicity” and Marmur proposed to use the term parahydrophobicity.57 This state can be predicted using the Cassie–Baxter equation and with multivalued roughness topographies such as overhangs, re-entrant structures, T-like structures or mushroom-like structures.58 Indeed, the air trapped below multiple roughness topographies can induce a negative Laplace pressure difference changing the liquid–vapor interface and impeding the liquid penetration.59 It is possible to control the liquid penetration inside the rough surfaces and have various adhesion strengths by adjusting the geometrical parameters of the different roughness topographies. Bormashenko and Starov also studied the liquid penetration inside capillaries of different sizes.60 They showed that small capillarities promote the Wenzel state while large capillarities favor the Cassie–Baxter state. In our case, for PProDOT(br-C4)2, the presence of the nanofibers as well as their low intrinsic hydrophobicity favored the Cassie–Baxter state but with an important liquid penetration inside the roughness, which led to a high water adhesion.
3.3 Influence of the number of deposition scans
Hence, due to the presence of the hard nanofibers, PProDOT(br-C4)2 was chosen to study the influence of the number of scans on the surface hydrophobicity and morphology. The influence of the number of scans on the surface hydrophobicity is represented in Fig. 5. Hence, similar hydrophobicities were obtained for one to three scans, whereas a decrease in θw was observed for five scans. Moreover, PProDOT(br-C4)2 was always sticky whatever the number of scans. The SEM images are given in Fig. 6. An increase in the characteristics of the nanofibers (diameter and length) was observed as a function of the numbers of scans.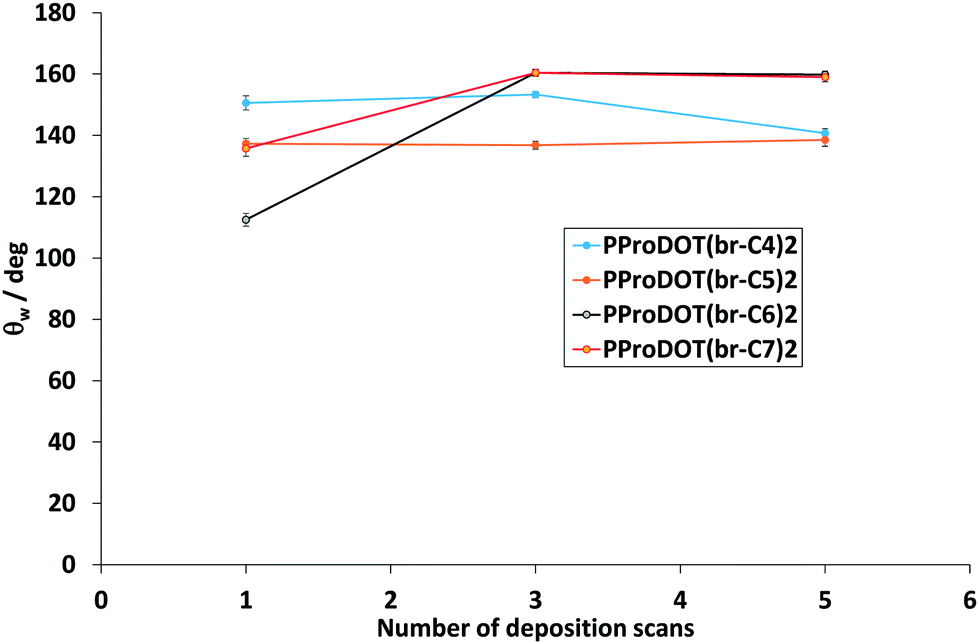 | ||
| Fig. 5 Apparent water contact angles of the polymers as a function of the deposition charge. | ||
 | ||
| Fig. 6 SEM images of PProDOT(br-C4)2, with a number of scans of (A) 1 and (B) 5; magnification: 25000×. | ||
As a consequence, the growth is not only in one dimension but is two-dimensional. To have more information, cross-section images were also performed as shown in Fig. 7. This figure confirms the vertical alignment of the nanofibers and the increase in the diameter and length with scan number. Moreover, for only one scan, most of the fibers were linear (Fig. 6A), but inclination angles were present after as shown in Fig. 7B. For five scans (Fig. 7C and D), the distance between the fibers seemed to be too important which may explain the decrease in θw.
 | ||
| Fig. 7 Cross-section SEM images of PProDOT(br-C4)2, with a number of scans of (A) 1, (B) 3 and (C and D) 5. | ||
4 Conclusions
This work allowed to advance in the formation of hard nanofibers using polymer materials. Indeed, they are often subjected to deformation after lateral collapse and coalescence, for example during evaporation, which is due to capillary forces and low stiffness of the fiber.46–49 Here, we have reported the synthesis and characterization of original ProDOT monomers containing branched alkyl chains for the generation of superhydrophobic films by cyclic voltammetry electrodeposition. These molecules indicate that hydrocarbon derivatives (cheaper, available as feed stocks, non-toxic) can be an alternative to long fluorocarbon chains (expensive, involved synthetic routes, bioaccumulable) to achieve anti-wetting properties. Surprisingly, the control of molecular shape and subsequent electrodeposition leads to superhydrophobic soft and also hard nanofibers. The decrease in the size of branched chains allowed to change the morphology from soft to hard nanofibers with a high increase in water adhesion due to a lower intrinsic hydrophobicity (sticky superhydrophobicity or parahydrophobicity). Moreover, in the case of the hard nanofibers, cross-section images revealed a vertical alignment of these fibers to the substrate. Furthermore, it was possible to control the height and the diameter of the hard nanofibers, as well as the distance between the fibers by the number the deposition scans. Such nanofibrous materials are extremely interesting and could be used for various biomedical applications such as in tissue engineering, biosensors or anti-bioadhesion. Such branched molecules offer new possibilities in materials chemistry to generate superhydrophobic nanofibers.Notes and references
- Y. Lai, L. Lin, F. Pan, J. Huang, R. Song, Y. Huang, C. Lin, H. Fuchs and L. Chi, Small, 2013, 9, 2945 CrossRef CAS PubMed.
- K. H. Lee, G. H. Kwon, S. J. Shin, J.-Y. Baek, D. K. Han, Y. Park and S. H. Lee, J. Biomed. Mater. Res., Part A, 2008, 90, 619 Search PubMed.
- M. J. Pérez-Roldan, A. Parracino, G. Ceccone, P. Colpo and F. Rossi, Plasma Processes Polym., 2014, 11, 577 CrossRef PubMed.
- F.-q. Sun, X.-s. Li, J.-k. Xu and P. t. Cao, Chin. J. Polym. Sci., 2010, 28, 705 CrossRef CAS PubMed.
- J. Tarrade, T. Darmanin, E. Taffin de Givenchy, F. Guittard, D. Debarnot and F. Poncin-Epaillard, Appl. Surf. Sci., 2014, 292, 782 CrossRef CAS PubMed.
- K. E. Park, K. Y. Lee, S. J. Lee and W. H. Park, Macromol. Symp., 2007, 249–250, 103 CrossRef PubMed.
- N.-N. Bui and J. R. McCutcheon, Environ. Sci. Technol., 2013, 47, 1761 CAS.
- J. E. Díaz, A. Barrero, M. Márquez and I. G. Loscertales, Adv. Funct. Mater., 2006, 16, 2110 CrossRef PubMed.
- H. Bellanger, T. Darmanin, E. Taffin de Givenchy and F. Guittard, Chem. Rev., 2014, 114, 2694 CrossRef CAS PubMed.
- E. Celia, T. Darmanin, E. Taffin de Givenchy, S. Amigoni and F. Guittard, J. Colloid Interface Sci., 2013, 402, 1 CrossRef CAS PubMed.
- M. Wolfs, T. Darmanin and F. Guittard, Polym. Rev., 2013, 53, 460 CrossRef CAS.
- T. Darmanin and F. Guittard, Prog. Polym. Sci., 2014, 39, 656 CrossRef CAS PubMed.
- W. Zhong, S. Liu, X. Chen, Y. Wang and W. Yang, Macromolecules, 2006, 39, 3224 CrossRef CAS.
- Y. Zhu, D. Hu, M. Wan, L. Jiang and Y. Wie, Adv. Mater., 2007, 19, 2092 CrossRef CAS PubMed.
- H. Fan, H. Wang, J. Guo, N. Zhao and J. Xu, J. Colloid Interface Sci., 2013, 409, 255 CrossRef CAS PubMed.
- Y. Zhu, J. Li, M. Wan and L. Jiang, Macromol. Rapid Commun., 2008, 29, 239 CrossRef CAS PubMed.
- J. Wang, J. Wang, Z. Wang and F. Zhang, Macromol. Rapid Commun., 2009, 30, 604 CrossRef CAS PubMed.
- N.-R. Chiou, C. Lu, J. Guan, L. J. Lee and A. J. Epstein, Nat. Nanotechnol., 2007, 2, 354 CrossRef CAS PubMed.
- S. B. Kulkarni, S. S. Joshi and C. D. Lokhande, Chem. Eng. J., 2011, 166, 1179 CrossRef CAS PubMed.
- H. Zhou, Z. Shi and Y. Lu, Synth. Met., 2010, 160, 1925 CrossRef CAS PubMed.
- J. Yan, X. Han, J. He, L. Kang, B. Zhang, Y. Du, H. Zhao, C. Dong, H. L. Wang and P. Xu, ACS Appl. Mater. Interfaces, 2012, 4, 2752 CAS.
- P. Xu, N. H. Mack, S. H. Jeon, S. K. Doorn, X. Han and H. L. Wang, Langmuir, 2010, 26, 8882 CrossRef CAS PubMed.
- Y. Zhu, J. Li, H. He, M. Wan and L. Jiang, Macromol. Rapid Commun., 2007, 28, 2230 CrossRef CAS PubMed.
- T. Darmanin, E. Taffin de Givenchy, S. Amigoni and F. Guittard, Adv. Mater., 2013, 25, 1378 CrossRef CAS PubMed.
- T. Darmanin and F. Guittard, J. Am. Chem. Soc., 2009, 131, 7928 CrossRef CAS PubMed.
- L. Xu, Z. Chen, W. Chen, A. Mulchandani and Y. Yan, Macromol. Rapid Commun., 2008, 29, 832 CrossRef CAS PubMed.
- H. Zhang, J. Wang, Z. Zhou, Z. Wang, F. Zhang and S. Wang, Macromol. Rapid Commun., 2008, 29, 68 CrossRef PubMed.
- H. Yan, K. Kurogi, H. Mayama and K. Tsujii, Angew. Chem., Int. Ed., 2005, 44, 3453 CrossRef CAS PubMed.
- J. Zang, C. M. Li, S. J. Bao, X. Cui, Q. Bao and C. Q. Sun, Macromolecules, 2008, 41, 7053 CrossRef CAS.
- C. Ding, Y. Zhu, M. Liu, L. Feng, M. Wan and L. Jiang, Soft Matter, 2012, 8, 9064 RSC.
- (a) S.-C. Luo, J. Sekine, B. Zhu, H. Zhao, A. Nakao and H.-h. Yu, ACS Nano, 2012, 6, 3018 CrossRef CAS PubMed; (b) S.-C. Luo, S. S. Liour and H.-h. Yu, Chem. Commun., 2010, 46, 4731 RSC.
- M. Wolfs, T. Darmanin and F. Guittard, Soft Matter, 2012, 8, 9110 RSC.
- P. Conte, T. Darmanin and F. Guittard, React. Funct. Polym., 2014, 74, 46 CrossRef CAS PubMed.
- T. Darmanin and F. Guittard, J. Am. Chem. Soc., 2011, 133, 15627 CrossRef CAS PubMed.
- O. Dunand, T. Darmanin and F. Guittard, ChemPhysChem, 2013, 14, 2947 CrossRef CAS PubMed.
- (a) M. Wolfs, T. Darmanin and F. Guittard, RSC Adv., 2013, 3, 647 RSC; (b) M. Wolfs, T. Darmanin and F. Guittard, Eur. Polym. J., 2013, 49, 2267 CrossRef CAS PubMed.
- T. Darmanin and F. Guittard, Mater. Chem. Phys., 2014, 146, 6 CrossRef CAS PubMed.
- T. Darmanin, C. Mortier and F. Guittard, Adv. Mater. Interfaces, 2014, 1, 1300094 Search PubMed.
- J. P. Giesy and K. Kannan, Environ. Sci. Technol., 2001, 35, 1339 CrossRef CAS.
- M. Houde, J. W. Martin, R. J. Letcher, K. R. Solomon and D. C. G. Muir, Environ. Sci. Technol., 2006, 40, 3463 CrossRef CAS.
- J. Eastoe, A. Paul, S. Nave, D. C. Steytler, B. H. Robinson, E. Rumsey, M. Thorpe and R. K. Heenan, J. Am. Chem. Soc., 2001, 123, 988 CrossRef CAS.
- A. Mohamed, K. Trickett, S. Y. Chin, S. Cummings, M. Sagisaka, L. Hudson, S. Nave, R. Dyer, S. E. Rogers, R. K. Heenan and J. Eastoe, Langmuir, 2010, 26, 13861 CrossRef CAS PubMed.
- Y. J. Kim, K. H. Park, J.-j. Ha, D. S. Chung, Y.-H. Kim and C. E. Park, Phys. Chem. Chem. Phys. 10.1039/c4cp00077c.
- C. Cui, Y. Sun, Z.-G. Zhang, M. Zhang, J. Zhang and Y. Li, Macromol. Chem. Phys., 2012, 213, 2267 CrossRef CAS PubMed.
- S. P. Mishra, K. Krishnamoorthy, R. Sahoo and A. Kumar, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 419 CrossRef CAS PubMed.
- A. J. Guenthner, S. Khombhongse, W. Liu, P. Dayal, D. H. Reneker and T. Kyu, Macromol. Theory Simul., 2006, 15, 87 CrossRef CAS PubMed.
- A. Grinthal, S. H. Kang, A. K. Epstein, M. Aizenberg, M. Khan and J. Aizenberg, Nano Today, 2011, 7, 35 CrossRef PubMed.
- X.-F. Wu and Y. A. Dzenis, Nanotechnology, 2007, 18, 285702 CrossRef.
- T. S. Kustandi, V. D. Samper, D. K. Yi, W. S. Ng, P. Neuzil and W. Sun, Adv. Funct. Mater., 2007, 17, 2211 CrossRef CAS PubMed.
- R. N. Wenzel, Ind. Eng. Chem., 1936, 28, 988 CrossRef CAS.
- A. B. D. Cassie and S. Baxter, Trans. Faraday Soc., 1944, 40, 546 RSC.
- T. Young, Philos. Trans. R. Soc. London, 1805, 95, 65 CrossRef.
- (a) L. Feng, Y. Zhang, J. Xi, Y. Zhu, N. Wang, F. Xia and L. Jiang, Langmuir, 2008, 24, 4114 CrossRef CAS PubMed; (b) J. B. K. Law, A. M. H. Ng, A. Y. He and H. Y. Low, Langmuir, 2014, 30, 325 CrossRef CAS PubMed.
- (a) S. Choo, H.-J. Choi and H. Lee, Matter. Lett., 2014, 121, 170 CrossRef CAS PubMed; (b) D. Sameoto and C. Menon, Smart Mater. Struct., 2010, 19, 103001 CrossRef.
- (a) Q. Cheng, M. Li, Y. Zheng, B. Su, S. Wang and L. Jiang, Soft Matter, 2011, 7, 5948 RSC; (b) Q. Cheng, M. Li, F. Yang, M. Liu, L. Li, S. Wang and L. Jiang, Soft Matter, 2012, 8, 6740 RSC.
- S. Yang, J. Ju, Y. Qiu, Y. He, X. Wang, S. Dou, K. Liu and L. Jiang, Small, 2014, 10, 294 CrossRef CAS PubMed.
- (a) A. Marmur, Soft Matter, 2012, 8, 6867 RSC; (b) A. Marmur, Soft Matter, 2013, 9, 7900 RSC; (c) A. Marmur, Langmuir, 2003, 19, 8343 CrossRef CAS.
- A. Marmur, Langmuir, 2008, 24, 7573 CrossRef CAS PubMed.
- J.-L. Liu, X.-Q. Feng, G. Wang and S.-W. Yu, J. Phys.: Condens. Matter, 2007, 19, 356002 CrossRef.
- (a) E. Bormashenko, Philos. Trans. R. Soc., A, 2010, 368, 4695 CrossRef CAS PubMed; (b) E. Bormashenko and V. Starov, Colloid Polym. Sci., 2013, 291, 343 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |