DOI:
10.1039/C4RA06646D
(Paper)
RSC Adv., 2014,
4, 44124-44129
Basicity-controlled synthesis of Li4Ti5O12 nanocrystals by a solvothermal method†
Received
4th July 2014
, Accepted 10th September 2014
First published on 10th September 2014
Abstract
Spinel-type lithium titanate (Li4Ti5O12) nanocrystals were synthesized at a low temperature with control of the basicity by a solvothermal method without any calcination processes. The Li–Ti–O precursor was produced under relatively high pH conditions (pH > 11) in the initial stage. The spinel phase was then formed through delithiation and dehydration from the precursor by lowering the pH of the system (pH < 10). The direct synthesis of the Li4Ti5O12 nanocrystals was achieved through self-control of the basicity using a two-phase system of water–ethanol and toluene–oleic acid. The Li4Ti5O12 crystals were formed from the precursor by decreasing the basicity. The average size of the nanocrystals prepared by the one-step method was ca. 13 nm, and the specific surface area was 158 m2 g−1. On the basis of the electrochemical measurement, the Li4Ti5O12 nanocrystals produced in a low-temperature solution system are applicable for an anode material of a lithium-ion rechargeable battery.
1. Introduction
In recent years, a wide variety of cathode and anode materials have been studied to improve the capacity and durability of rechargeable lithium-ion batteries. Spinel lithium titanate (Li4Ti5O12) is a stable anode material having a reaction potential of 1.55 V (vs. Li/Li+) and a theoretical capacity of 175 mA h g−1 for lithium-ion batteries. This oxide is well known as zero-strain insertion material having high durability against the insertion–desertion of charge–discharge cycles.1–4 The size reduction of the crystalline particles is important for further improvement of the capacity and rate performance through expansion of the interface area for the electrochemical reaction and shortening of the distance for the lithium-ion diffusion.5 Thus, the nanosized particles, hierarchical structures and mesoporous frameworks of Li4Ti5O12 were reported to enhance the performance.6–11 However, calcination processes above 400 °C are required to produce the spinel structure, and their nanostructures would be degraded at high temperatures.8–16 In the current work, we directly synthesized the nanocrystals of Li4Ti5O12 in a solution system by controlling the basicity without any subsequent calcination processes.
Hydrothermal and solvothermal methods are useful for the synthesis of nanocrystals and hierarchical structures of various metal oxides.17–23 However, they generally have been hindered by problems such as controlling their chemical composition, size distribution and crystallinity. Particularly, exploration of the optimum conditions for the synthesis of complex oxides is required. In previous works, alkali-earth titanium oxides having a stable perovskite structure (BaTiO3, SrTiO3, and CaTiO3) and layered alkali titanium oxides (Na2Ti6O13, KTi8O16.5, and CsxTi2−x/4□x/4O4) were prepared through hydrothermal routes by adjusting conditions including temperature and pH.24–29 Although Li2TiO3 was obtained by a hydrothermal process, the preparation of crystalline spinel Li4Ti5O12 has not been achieved without subsequent calcination.30,31 While the diffraction pattern assignable to Li4Ti5O12 was reported for a product obtained through a solvothermal process by Fattakhova et al., the structural analysis was insufficient and the preparation condition was ambiguous.32 In recent papers, formation of the Li4Ti5O12 phase was achieved through a nonaqueous or supercritical synthesis without an annealing process.33,34 However, these methods required restricted conditions such as an inert atmosphere or high temperature and high pressure. Moreover, the Li4Ti5O12 prepared by the nonaqueous synthesis included TiO2 as an impurity, or Li2TiO3 appeared after annealing. Therefore, development of the aqueous-solution system for preparation of spinel-type Li4Ti5O12 is still important to produce its nanometric structure with a simple technique for various applications.
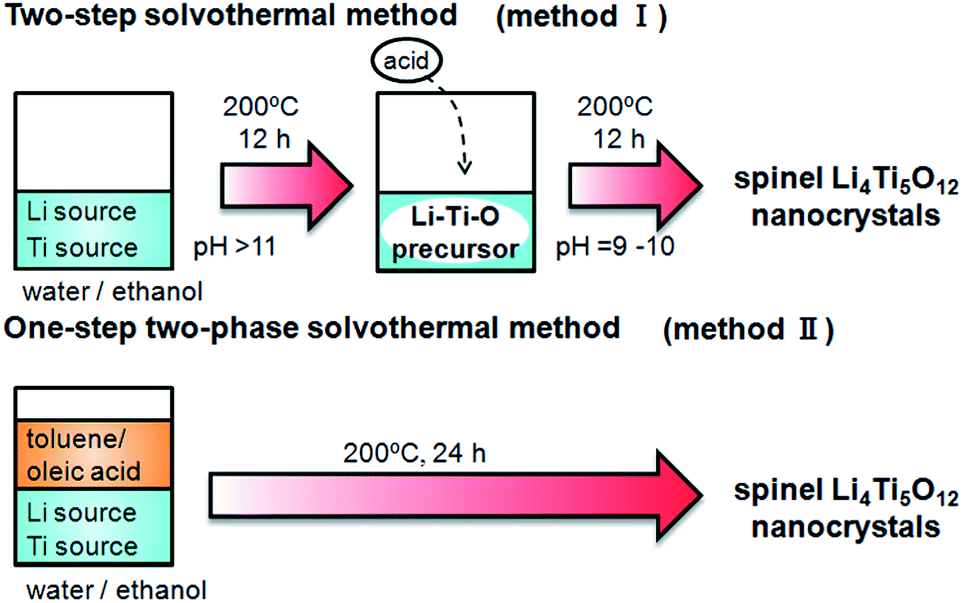
In present study, single-phase Li4Ti5O12 nanocrystals were directly obtained by a two-step solvothermal method (method I) or a one-step two-phase method (method II) as shown in Scheme 1. The basicity control after formation of the Li–Ti–O precursor was found to be essential for synthesis of the spinel crystal. The resultant Li4Ti5O12 crystals, ca. 13 nm in diameter, exhibited a good capacity as an anode material for lithium-ion batteries even without heat treatment. The cycle stability of the nanocrystals was greatly enhanced through improvement of the crystallinity by calcination. The basicity-controlled solvothermal technique would be useful for preparation of a wide variety of complex oxide materials.
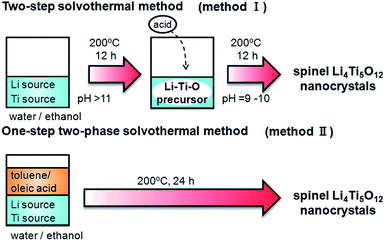 |
| | Scheme 1 Solvothermal methods for basicity-controlled syntheses of the Li4Ti5O12 nanocrystals. | |
2. Experimental section
The pH-controlled two-step solvothermal method (method I in Scheme 1) was examined as follows. All the reagents in the present work were used without further purification. A 1.0 mol dm−3 LiOH solution was prepared by dissolving LiOH·H2O (Kanto Chemical) in 20 cm3 of a mixture of purified water and ethanol (1/1 volume). Bis(ammonium lactate) titanium dihydroxide (Sigma-Aldrich) was mixed into the solution (molar ratio of Li/Ti: 10), and a white precipitate formed immediately. The first solvothermal process was carried out in a Teflon-lined stainless steel autoclave at 200 °C for 12 h. After cooling to room temperature, the pH of the mixture was decreased to a value between 9 and 10 by the addition of 0.014 mol of acetic acid. The second solvothermal process was then carried out at 200 °C for 12 h. The obtained precipitates were washed by ethanol and purified water and dried at 60 °C in air.
We directly prepared Li4Ti5O12 crystals using a basicity-controlled two-phase solvothermal method (method II in Scheme 1). The same mixture of purified water and ethanol containing LiOH·H2O and bis(ammonium lactate) titanium dihydroxide as for the two-step method was used for the one-step two-phase method. Toluene (20 cm3) containing 32 mmol of oleic acid (Wako Pure Chemical) and tert-butylamine (Wako Pure Chemical) was carefully poured to the water–ethanol mixture. The solvothermal treatment of the two-phase system was carried out at 200 °C for 12 or 24 h. The obtained precipitates were washed by ethanol and purified water and dried at 60 °C in air.
The crystal phases were identified by X-ray diffraction (XRD: Rigaku MiniFlex II and Bruker D8 Advance) with CuKα radiation. Crystalline domain sizes D(hkl) are calculated by the Scherrer equation. The values of full width at half maximum (FWHM) were calibrated with a highly crystalline standard sample. The morphologies of particles were observed by a transmission electron microscope (TEM: FEI TECNAI-F20) operated at 200 kV. The specific surface area was measured by the N2-adsorption/desorption measurement (Shimazu Trister 3000). The electrochemical performance was measured with a beaker-type three-electrode cell. The working electrode was a slurry composed of 80 wt% Li4Ti5O12 or other Li–Ti–O products, 10 wt% acetylene black carbon and 10 wt% polyvinylidene difluoride binder dispersed in N-methyl pyrrolidinone. The slurry was dropped on a Ni mesh and dried at 80 °C in vacuum. A lithium metal was used for the counter and reference electrodes on a Ni mesh. The electrolyte was 1 mol dm−3 of LiClO4 in ethylene carbonate and diethyl carbonate (1/1 volume). The cell was assembled in an argon-filled glove box. Galvanostatic discharge–charge measurements were performed in a potential range between 1.0 and 2.7 V vs. Li/Li+ at room temperature.8
3. Results and discussion
3.1. Basicity-controlled two-step solvothermal method (method I)
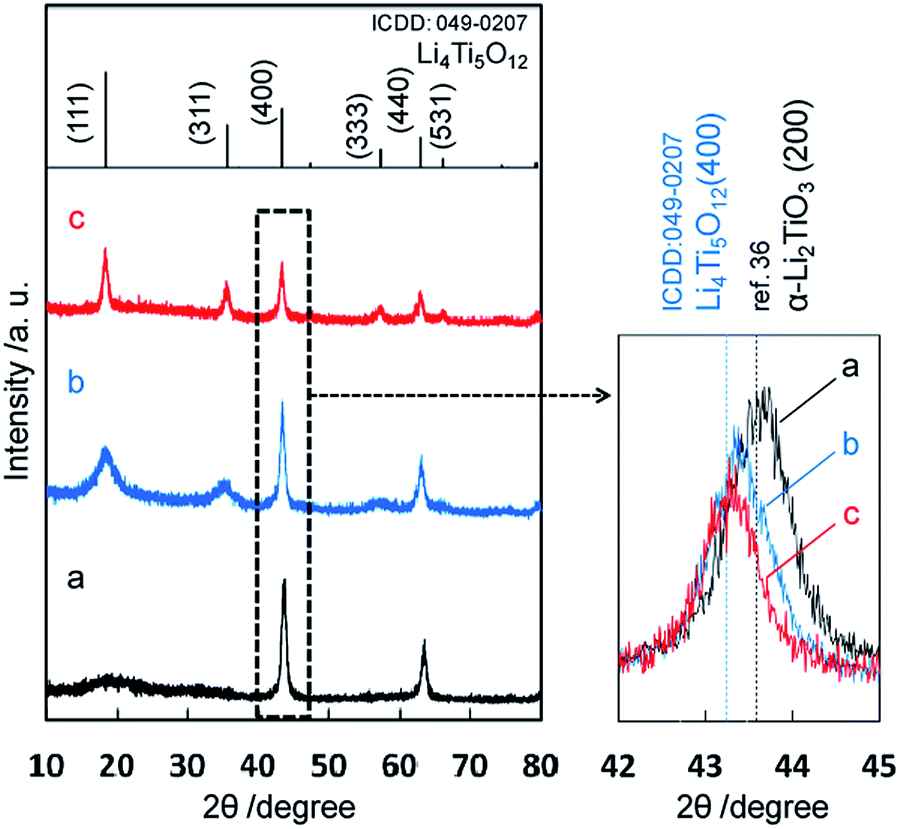
The precipitate was obtained through the solvothermal reaction with lithium and titanium ions at a pH value over 11 in the first step. According to the XRD pattern (Fig. 1a), the product of the first step was the Li–Ti–O precursor, which was reported in previous works for hydrothermal synthesis of lithium titanates.8,9,13,14,34–36 The Li–Ti–O precursor was deduced to be nominal cubic α-Li2TiO3 due to the following reasons. The diffraction pattern of the intermediate phase was very similar to the XRD profile assigned to α-Li2TiO3 by Laumann et al.36 Basically, two major diffraction peaks of (200) and (220) appear for α-Li2TiO3 in the range of 2θ = 10–80°. The spacings for d(200) of α-Li2TiO3, d(−133) of monoclinic β-Li2TiO3, and d(400) of cubic Li4Ti5O12 are 0.2075 nm (ref. 36), 0.2075 nm (ICDD: 033-0831), and 0.2091 nm (ICDD: 049-0207), respectively. As shown in Fig. 1, the diffraction peak at 2θ = 43.7° from the intermediate phase was corresponding to 0.2075 nm. This result supports that the Li–Ti–O precursor was not Li4Ti5O12. Several diffraction peaks (002), (200), (006) and (062) assigned to β-Li2TiO3 were not observed for the Li–Ti–O precursor. Thus, the intermediate phase was deduced to be nominal α-Li2TiO3, namely some lithium defects in the α-Li2TiO3.
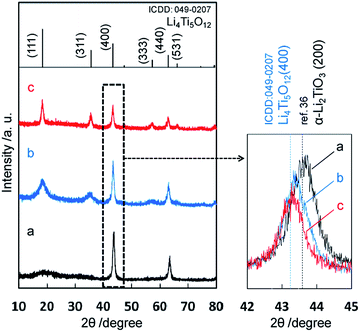 |
| | Fig. 1 XRD patterns of precipitates obtained by method I. The precursor obtained in the first stage (a), the final product in the second step (b), and the final product after calcination at 500 °C for 3 h (c). The top of this figure shows a diffraction pattern of Li4Ti5O12 (ICDD: 049-0207). | |
The diffraction peaks of the final product after the second solvothermal step at a relatively low pH value (9–10) agreed with those of spinel Li4Ti5O12, although the diffraction bands corresponding to the (111) and (311) planes were broadened (Fig. 1b). The diffraction peak position (2θ = 43.3°) of the final product, which corresponded to the spinel Li4Ti5O12, shifted from that of the Li–Ti–O precursor (2θ = 43.7°). Moreover, the diffraction peaks of (020), (110), (−111), and (021) for β-Li2TiO3 were not observed in the range of 2θ = 20–22°. Thus, we succeeded in the production of spinel Li4Ti5O12 from the Li–Ti–O precursor by the solvothermal method without subsequent heat treatment.
Table 1 shows various sizes of the obtained Li4Ti5O12 samples. The table includes the FWHM of the diffraction peaks, the crystalline domain sizes, D(111) and D(400), the particle sizes DTEM estimated by TEM, and the particle sizes DBET calculated from specific surface area, DBET = 6/(SBETρ). ρ is the density of the Li4Ti5O12. The weak (111) signal of Li4Ti5O12 is deduced to be a lattice distortion in the [111] direction. In the second solvothermal step, Li4Ti5O12 was formed from α-Li2TiO3 which was produced in the first step. The (400) plain of Li4Ti5O12 is inherited from the (200) plain of cubic α-Li2TiO3 because of the agreement of their d-spacings. On the other hand, the (111) plane of Li4Ti5O12 would be gradually formed because no similar planes exist in the precursor phase. Thus, we observed a strong signal due to the highly ordered (400) plane and a relatively weak signal due to the disordered (111) plane.
Table 1 FWHM of the diffraction peaks, the crystalline domain sizes D(hkl), the particle sizes DTEM observed by TEM, the particle sizes DBET calculated from specific surface area, and the specific surface areas SBET for obtained Li4Ti5O12 samples. DBET = 6/(SBETρ). ρ is the density of the Li4Ti5O12
| |
FWHM(111)/degree |
FWHM(400)/degree |
D(111)/nm |
D(400)/nm |
DTEM/nm |
DBET/nm |
SBET/m2 g−1 |
| These values are affected by the lattice distortion. |
| Method I |
2.97 |
0.71 |
2.7a |
13.0 |
11.7 (4.9) |
10.8 |
159.1 |
| Method II |
2.07 |
0.72 |
3.9a |
12.7 |
13.0 (4.1) |
10.9 |
157.9 |
| Method I calcined |
0.83 |
0.63 |
10.2 |
14.9 |
11.7 (3.3) |
12.7 |
134.8 |
| Method II calcined |
0.67 |
0.67 |
13.0 |
13.8 |
13.9 (4.9) |
17.1 |
100.5 |
Fig. 2 shows TEM images of the products. Round-shaped particles with a diameter of ca. 13 nm were observed for the Li–Ti–O precursor (Fig. 2a). Almost the same sized particles of Li4Ti5O12 crystal were obtained by the second step. The lattice fringes corresponding to the (111) plane of Li4Ti5O12 support that the crystalline nanoparticles were generated by the second step (Fig. 2b). The crystalline domain sizes calculated by the Scherrer equation are D(111) = 2.7 nm (as-prepared sample), D(400) = 13.0 nm (as-prepared sample), D(111) = 10.2 nm (calcined sample), and D(400) = 14.9 nm (calcined sample). The value of D(400) = 13.0 nm for the as-prepared sample was almost corresponded to the particles size DTEM = 11.7 nm observed by TEM. On the other hand, the value of D(111) was smaller than DTEM. The difference suggests the presence of a lattice distortion in the [111] direction. Because D(111) was clearly increased by the calcination, the crystallinity of the calcined sample is better than that of the as-prepared sample.
 |
| | Fig. 2 TEM images of the precipitates obtained by method I. The precursor obtained in the first stage (a), the final product in the second step (b), and the final product after calcination at 500 °C for 3 h (c). The insets in (b) and (c) show the lattice fringes of the (111) plane of Li4Ti5O12 (d(111) = 0.48 nm). | |
Here we discuss the formation mechanism of the spinel phase under the solvothermal conditions (Formulas (1)–(3)). Amorphous titanium dioxide was precipitated through a hydrolysis of bis(ammonium lactate) titanium dihydroxide under a basic condition before the first solvothermal step. The Li–Ti–O precursor, nominal α-Li2TiO3, then formed under a basic condition over pH 11 in the first solvothermal step. In the previous study, the transformation of the Li–Ti–O precursor to Li4Ti5O12 requires heat treatment at temperatures above 400 °C. On the other hand, α-Li2TiO3 is delithiated even at room temperature in pure water or an acid solution.14,36 Thus, (Li2−xHx)TiO3 could be formed through delithiation of the precursor by lowering the pH to ∼10 by the addition of acetic acid in the second step. Finally, the spinel Li4Ti5O12 is produced by dehydration of (Li2−xHx)TiO3. (x = 1.2 is confirmed for formation of the spinel Li4Ti5O12). When the pH value was decreased to below pH 9 by the addition of a large amount of acetic acid, we observed the formation of anatase TiO2 through H2TiO3 due to excess delithiation. This fact supports the formation mechanism of the spinel phase in the solution system proposed here. When hydrochloric acid or nitric acid was used as a pH controller instead of acetic acid, spinel Li4Ti5O12 was also produced. These results support the role of the acid in the reaction.
| | |
2LiOH + TiO2 → Li2TiO3 + H2O (>pH 11)
| (1) |
| | |
Li2TiO3 + xH+ → (Li2−xHx)TiO3 + xLi+ (delithiation)
| (2) |
| | |
(Li2−xHx)TiO3 → Li2−xTiO3−x/2 + x/2H2O (dehydration)
| (3) |
3.2. Basicity-controlled one-step two-phase method (method II)
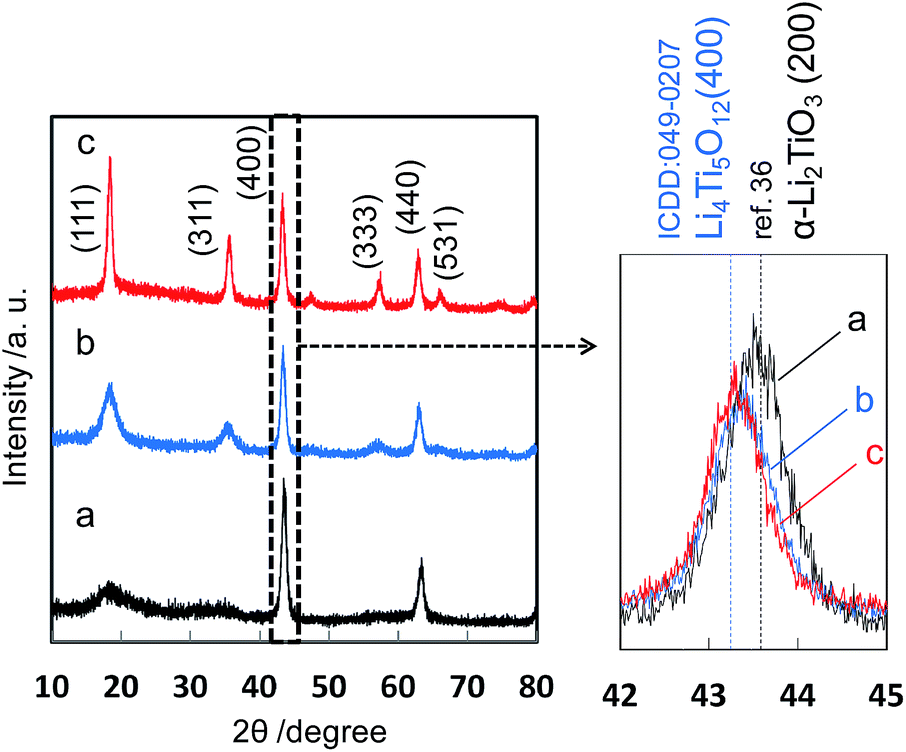
Based on the reaction process of the two-step solvothermal method mentioned above, a delithiation process induced by decreasing the basicity is essential for the formation of the spinel Li4Ti5O12 phase in the solution system. Next we developed the one-pot synthesis of the spinel Li4Ti5O12 nanocrystal with self-control of the basicity using the two-phase system of water–ethanol and toluene–oleic acid. As shown in Fig. 3, the pure spinel phase was directly obtained after the solvothermal reaction at 200 °C for 24 h, whereas the Li–Ti–O precursor was observed for 12 h. The Li–Ti–O precursor was produced under a relatively high pH condition due to the dissolution of lithium hydroxide in the initial stage. The Li4Ti5O12 crystals were then formed from the precursor by decreasing the basicity in the progressive stage. This means that a gradual transfer of a certain amount of the organic acid from the toluene phase lowers the basicity of the water phase by disassociation. tert-Butylamine was needed for the two-phase solvothermal process because a slight amount of anatase TiO2 as an impurity formed without using tert-butylamine. The presence of tert-butylamine may support decreasing the pH stably or stabilizing titanium hydroxide ions as dissolution species. Additionally, the formation of the Li4Ti5O12 nanocrystal required a solvothermal temperature equal to or higher than 180 °C (Fig. S2†). The particle size and the specific surface area of the products were ca. 13 nm (Fig. 4) and 158 m2 g−1, respectively. The crystallinity was improved by calcination at 500 °C with a slight increase in the particle size.
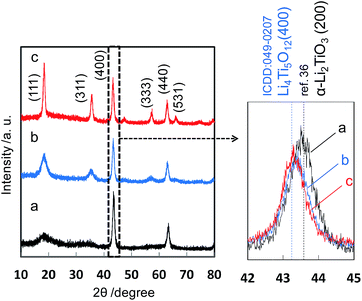 |
| | Fig. 3 XRD patterns of the precursor obtained by method II in the initial stage (12 h) (a), the Li4Ti5O12 crystals in the progressive stage (24 h) (b), and after calcination at 500 °C for 3 h (c). | |
 |
| | Fig. 4 TEM images of the Li4Ti5O12 crystals prepared by method II (a) and after calcination at 500 °C for 3 h (b). | |
3.3. Electrochemical analyses of Li4Ti5O12 nanocrystals
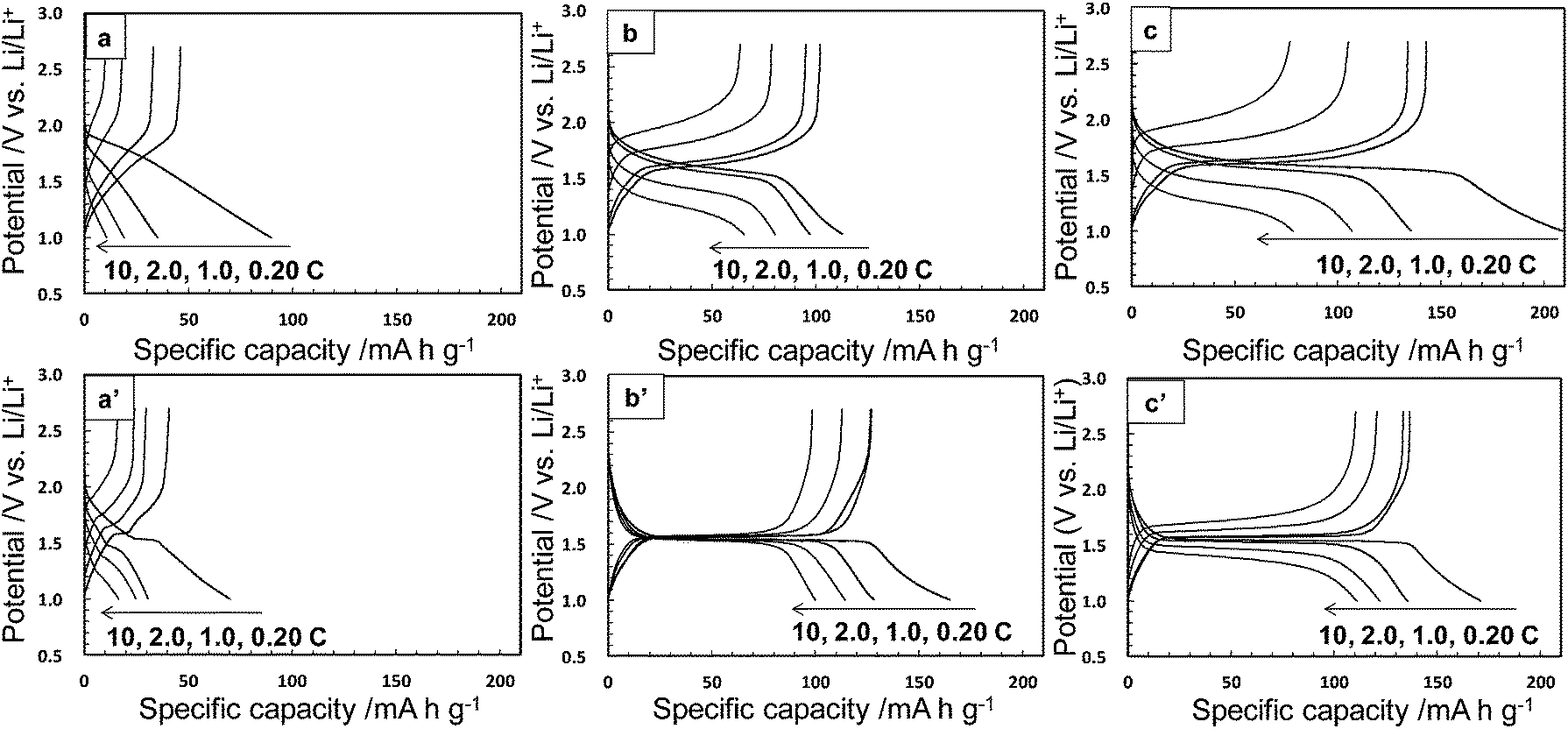
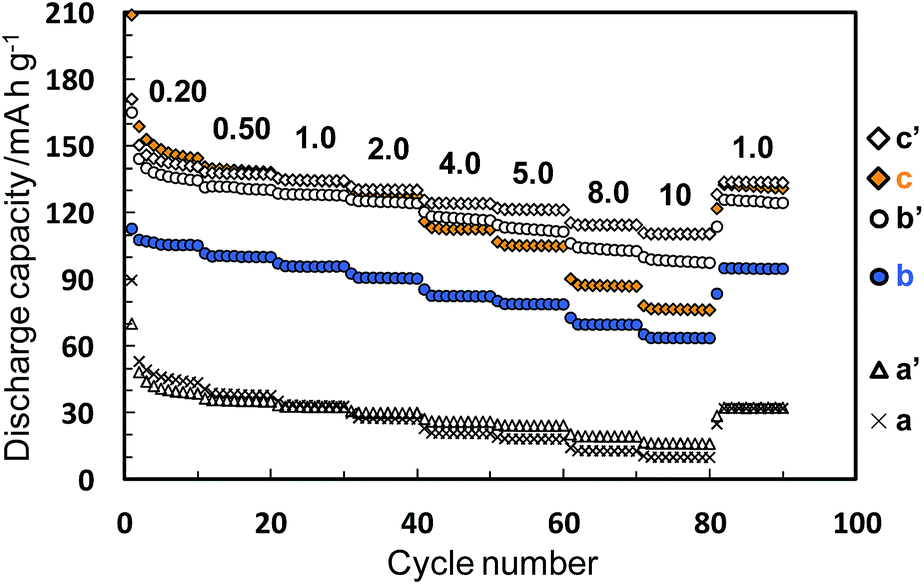
The electrochemical performance was measured for the Li–Ti–O precursor and Li4Ti5O12 nanocrystals obtained through methods I and II. The samples calcined at 500 °C were also characterized under the same conditions. Fig. 5 and 6 show charge–discharge curves and the cycle and rate performances, respectively, at several current rates in the voltage between 1.0 and 2.7 V. We observed unclear voltage plateaus and low capacity values for the Li–Ti–O precursor (a) and the calcined one (a′). On the other hand, the Li4Ti5O12 nanocrystals obtained by method I (b) showed a reaction voltage plateau around 1.55 V (vs. Li/Li+), and the first discharge and reversible capacity were 113 and 102 mA h g−1, respectively, at 0.20 C (1.0 C = 175 mA g−1). The first reversible capacity of the nanocrystals was increased to 128 mA h g−1 by the calcination (b′). The capacity of the Li4Ti5O12 crystals obtained by method II (c) was 143 mA h g−1, which was even higher than that of the calcined one (c′) (137 mA h g−1) and was comparable to the results of well-crystallined Li4Ti5O12 reported in a previous article.15 These results indicate that the Li4Ti5O12 nanocrystals obtained directly by method II showed a good capacity even without heat treatment. The capacity value of as-prepared Li4Ti5O12 nanocrystals (b and c) was decreased by increasing the rate current. On the other hand, the performance of the Li4Ti5O12 nanocrystals was improved by the subsequent calcination (b′ and c′). A high crystallinity would be required for a fast electrode reaction at a high rate. Because the difference in the crystallinity of the calcined samples is smaller than that of the as-prepared samples, the difference in reversible capacity of the calcined samples obtained method I and II was decreased. According to the difference of FWHM(111) (2.97° (method I) and 2.07° (method II)), the crystallinity of the as-prepared sample of method II was higher than that of method I. We think that the performance was not affected by tert-butylamine because the organic molecules were easily removed in the washing process. In the one-step process, the Li4Ti5O12 nanocrystals are directly formed through the transformation of the fresh precursor phase in the solution system. Thus, the higher crystalline products are easily obtained by this simple technique. At a low current rate (0.20–1.0 C), the performance of as-prepared Li4Ti5O12 nanocrystals by method II (c) was higher than that of the calcined one. The higher performance of the Li4Ti5O12 nanocrystals may be attributed to the coexistence of the high crystallinity and the high specific surface area due to loose aggregation without the calcination process. This suggests the advantage of direct synthesis of Li4Ti5O12 nanocrystals by the solvothermal method. Tailing was observed in the first discharge profiles (0.20 C). According to ref. 34, the irreversible capacity was ascribed to the surface reaction of nanosized Li4Ti5O12 samples. Therefore, the origin of tailing in the first discharge curves for our samples would be attributed to the presence of a high surface area originating form nanosized Li4Ti5O12 particles. In the previous works, the electrochemical performance of as-prepared sample obtained by the solvothermal method was quite lower than that of calcined samples due to their lower crystallinity. Highly crystalline products can be prepared even through the low-temperature process by adjusting the preparation conditions. The direct synthesis of the products exhibiting a high performance would enable a low-temperature technique which is needed such for low energy consumption process, fabrication of nanoscale textures, and surface modification with organic molecules.
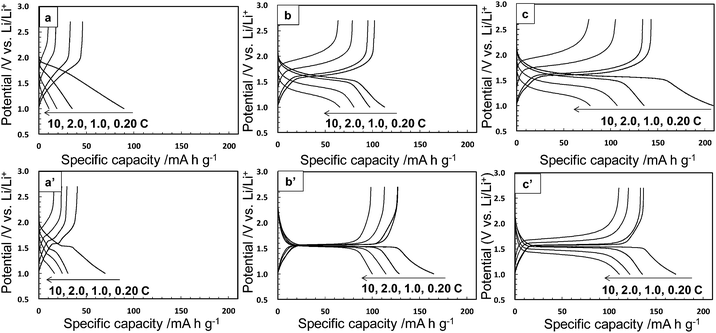 |
| | Fig. 5 Charge–discharge curves at various current rates (1.0 C = 175 mA g−1) of the Li–Ti–O precursor (a), the Li4Ti5O12 nanocrystals obtained by methods I and II (b and c, respectively), and the calcined samples at 500 °C for 3 h (a′, b′, and c′). | |
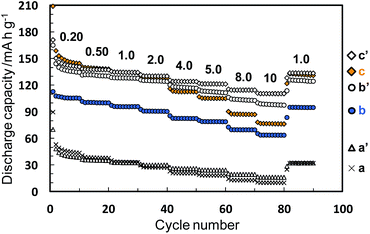 |
| | Fig. 6 Rate and cycle performances of the Li–Ti–O precursor (a), the Li4Ti5O12 nanocrystals obtained by methods I and II (b and c, respectively), and the calcined samples at 500 °C for 3 h (a′, b′ and c′). The numbers above the plots indicate current rates per C (1.0 C = 175 mA g−1). | |
4. Conclusions
Spinel Li4Ti5O12 nanocrystals were produced through the pH-controlled two-step solvothermal process. We found that the formation of Li4Ti5O12 crystals requires the nucleation of the Li–Ti–O precursor under a high pH condition in the initial stage and its transformation with a decrease of pH in the progressive stage of the solvothermal reaction. Then we succeeded in the direct synthesis of spinel Li4Ti5O12 nanocrystals by the one-step two-phase solvothermal process using a toluene–oleic acid system as a novel basicity-control method. The Li4Ti5O12 nanocrystals of ca. 13 nm in diameter prepared by the one-step two-phase solvothermal process showed a high specific capacity for application as an anode material.
Acknowledgements
This work was partially supported by the Advanced Low Carbon Technology Research and Development Program of Strategic Basic Research Programs from Japan Science and Technology Agency.
Notes and references
- T. Ohzuku, A. Ueda and N. Yamamoto, J. Electrochem. Soc., 1995, 142, 1431–1435 CrossRef CAS PubMed.
- K. M. Colbow, J. R. Dahn and R. R. Haering, J. Power Sources, 1989, 26, 397–402 CrossRef CAS.
- K. Zaghib, M. Simoneau, M. Armand and M. Gauthier, J. Power Sources, 1999, 81–82, 300–305 CrossRef CAS.
- F. Ronci, P. Reale, B. Scrosati, S. Panero, V. R. Albertini, P. Perfetti, M. di Michiel and J. M. Merino, J. Phys. Chem. B, 2002, 106, 3082–3086 CrossRef CAS.
- H.-G. Jung, S.-T. Myung, C. S. Yoon, S.-B. Son, K. H. Oh, K. Amine, B. Scrosati and Y.-K. Sun, Energy Environ. Sci., 2011, 4, 1345–1351 CAS.
- L. Kavan and M. Grätzel, Electrochem. Solid-State Lett., 2002, 5, A39–A42 CrossRef CAS PubMed.
- A. S. Prakash, P. Manikandan, K. Ramesha, M. Sathiya, J.-M. Tarascon and A. K. Shukla, Chem. Mater., 2010, 22, 2857–2863 CrossRef CAS.
- C. Jiang, E. Hosono, M. Ichihara, I. Honma and H. Zhou, J. Electrochem. Soc., 2008, 155, A553–A556 CrossRef CAS PubMed.
- L. Shen, C. Yuan, H. Luo, X. Zhang, K. Xu and Y. Xia, J. Mater. Chem., 2010, 20, 6998–7004 RSC.
- Y. Tang, L. Yang, Z. Qui and J. Huang, J. Mater. Chem., 2009, 19, 5980–5984 RSC.
- Y.-S. Lin and J.-G. Duh, J. Power Sources, 2011, 196, 10698–10703 CrossRef CAS PubMed.
- D. Fattakhova and P. Krtil, J. Electrochem. Soc., 2002, 149, A1224–A1229 CrossRef CAS PubMed.
- A. Zhang, Z. M. Zheng, F. Y. Cheng, Z. L. Tao and J. Chen, Sci. China: Chem., 2011, 54, 936–940 CrossRef CAS.
- J. Lu, C. Nan, Q. Peng and Y. Li, J. Power Sources, 2012, 202, 246–252 CrossRef CAS PubMed.
- H. Yan, Z. Zhu, D. Zhang, W. Li and Qilu, J. Power Sources, 2012, 219, 45–51 CrossRef CAS PubMed.
- Y. F. Tang, L. Yang, Z. Qiu and J. S. Huang, Electrochem. Commun., 2008, 10, 1513–1516 CrossRef CAS PubMed.
- H. Cheng, J. Ma, Z. Zhao and L. Qi, Chem. Mater., 1995, 7, 663–671 CrossRef CAS.
- Q. Wu, X. Chen, P. Zhang, Y. Han, X. Chen, Y. Yan and S. Li, Cryst. Growth Des., 2008, 8, 3010–3018 CAS.
- V. Subramanian, H. Zhu, R. Vajtai, P. M. Ajayan and B. Wei, J. Phys. Chem. B, 2005, 109, 20207–20214 CrossRef CAS PubMed.
- H.-X. Mai, L.-D. Sun, Y.-W. Zhang, R. Si, W. Feng, H.-P. Zhang, H.-C. Liu and C.-H. Yan, J. Phys. Chem. B, 2005, 109, 24380–24385 CrossRef CAS PubMed.
- C.-T. Dinh, T.-D. Nguyen, F. Kleitz and T.-O. Do, ACS Nano, 2009, 3, 3737–3743 CrossRef CAS PubMed.
- F. Lu, W. Cai and Y. Zhang, Adv. Funct. Mater., 2008, 18, 1047–1056 CrossRef CAS.
- G. Tian, Y. Chen, W. Zhou, K. Pan, Y. Dong, C. Tian and H. Fu, J. Mater. Chem., 2011, 21, 887–892 RSC.
- A. Testino, M. T. Buscaglia, V. Buscaglia, M. Viviani, C. Bottino and P. Nanni, Chem. Mater., 2004, 16, 1536–1543 CrossRef CAS.
- M.-H. Um and H. Kumazawa, J. Mater. Sci., 2000, 35, 1295–1300 CrossRef CAS.
- W. Dong, G. Zhao, B. Song, G. Xu, J. Zhou and G. Han, CrystEngComm, 2012, 14, 6990–6997 RSC.
- D.-S. Seo, H. Kim and J.-K. Lee, J. Cryst. Growth, 2005, 275, e2371–e2376 CrossRef PubMed.
- C.-Y. Xu, Q. Zhang, H. Zhang, L. Zhen, J. Tang and L.-C. Qin, J. Am. Chem. Soc., 2005, 127, 11584–11585 CrossRef CAS PubMed.
- N. Masaki, S. Uchida and T. Sato, J. Mater. Chem., 2002, 12, 305–308 RSC.
- M. Tomiha, N. Masaki, S. Uchida and T. Sato, J. Mater. Sci., 2002, 37, 2341–2344 CrossRef CAS.
- M. Sugita, M. Tsuji and M. Abe, Bull. Chem. Soc. Jpn., 1990, 63, 1978–1984 CrossRef CAS.
- D. Fattakhova, V. Petrykin, J. Brus, T. Kostlánová, J. Dědeček and P. Krtil, Solid State Ionics, 2005, 176, 1877–1885 CrossRef CAS PubMed.
- S.-H. Yu, A. Pucci, T. Herntrich, M.-G. Willinger, S.-H. Baek, Y.-E. Sung and N. Pinna, J. Mater. Chem., 2011, 21, 806–810 RSC.
- A. Laumann, M. Bremholm, P. Hald, M. Holzapfel, K. T. Fehr and B. B. Iversen, J. Electrochem. Soc., 2012, 159, A166–A171 CrossRef CAS PubMed.
- A. Laumann, K. M. Ø. Jensen, C. Tyrsted, M. Bremholm, K. T. Fehr, M. Holzapfel and B. B. Iversen, Eur. J. Inorg. Chem., 2011, 2221–2226 CrossRef CAS.
- A. Laumann, K. T. Fehr, M. Wachsmann, M. Holzapfel and B. B. Iversen, Solid State Ionics, 2010, 181, 1525–1529 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1 and S2. See DOI: 10.1039/c4ra06646d |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a