
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA hydroquinone based palladium catalyst for room temperature nitro reduction in water†
Alok Kumar‡
,
Kallol Purkait‡,
Suman Kr. Dey,
Amrita Sarkar and
Arindam Mukherjee*
Department of Chemical Sciences, Indian Institute of Science Education and Research Kolkata, Mohanpur Campus, Mohanpur-741246, India. E-mail: a.mukherjee@iiserkol.ac.in; Fax: +91 33 25873020
First published on 13th August 2014
Abstract
A hydroquinone based palladium complex [Pd(H2L)(Cl)2] (1), acts as an efficient room temperature catalyst for reduction of nitroarenes in water as solvent. 1 also acts as a tandem catalyst for Suzuki–Miyaura cross coupling in ethanol followed by reduction of nitroarenes in one pot with a loading of 0.25 mol% catalyst.
Palladium based catalysts are well known in the literature for their efficiency in a wide spectrum of coupling reactions and reduction of various functionalities.1 Such catalytic reactions are of importance for their use in industrial applications spanning from pharmaceuticals to polymers or syntheses of natural products, dyes.2 Aryl amine derivatives are very important intermediates in syntheses of many of the above using nitroarene precursors.3 Room temperature reduction of nitroarenes is advantageous since there are compounds which are susceptible to degradation when heated during reduction. Several transition metal based catalysts are known to perform reduction of nitroarenes.4 Most metal catalyzed nitro reduction reactions require higher temperatures,4f,5 hydrogen gas,3b,6 or use of expensive hydride source like silanes.4b,5e,g,7 Among the most recent ones the work of Beller and co-workers used a Fe–phenanthroline catalyst (1.0 mol%) at 100 °C in THF with four molar equivalent hydrazine as the hydrogen source for efficient and very selective reduction of nitroarenes.5b However, metal complexes that reduces nitroarenes at room temperature are scarce (Table 1). Most of such catalysts are nano particles (Table 1). Hence performing the nitroarene reduction in a green solvent like water at room temperature with a discrete metal complex is challenging. Very few catalysts perform the reduction of nitroarenes at room temperature with water as the solvent (Table 1) viz. Zn-dust (10 eq.) in surfactant (TPGS-750 M) water mixture8 and the ionic liquid based PVDF–[C6(mpy)2][NiCl4]22 (5 mol%) catalyst9 are needed in less amounts, are less toxic to environment are of high interest for their potential applications.10 Such catalysts are air stable, efficient, economically viable and hence greener. The hydroquinone ligand used in this work is synthesized in a single step and is stable. The Palladium catalyst is active in lesser mol% without a phosphine or carbene ligand. To the best of our knowledge the use of hydroquinone in nitroarene reduction is not known.
| Sl. no. | Catalyst | t | mol% | H-source and other additive | Solvent | Ref. |
|---|---|---|---|---|---|---|
| a TMDS = 1,1,3,3-tetramethyldisiloxane.b Argon atmosphere.c NAP = Nano Active™ Magnesium Oxide Plus.d 75 mg per mol of substrate.e Nitrogen atmosphere.f Nanocomposite.g Used 20 mg mol−1 of substrate.h PAMAM–PVAm/SBA-15 = polyamidoamine–polyvinylamine/mesoporous silica catalyst-0.1 g mol−1 of substrate.i PHMS = polymethylhydro-siloxane.j PVDF = polyvinylidene fluoride, mpy = 3-methylpyridinium.k 10 equivalent.l TPGS-750 M = DL-α-tocopherol methoxypolyethylene glycol succinate solution. | ||||||
| 1 | Fe-NPs | 2–3 h | 300 | Water | Water | 4a |
| 2 | Au-NPs | 10 min to 24 h | 0.5 | TMDSa (4.5 eq.)b | Ethanol | 4b |
| 3 | NAP-Mg–Au(0)c | 1 min to 4 h | d | NaBH4 (47 eq.)e | Water | 4d |
| 4 | Pd/C | 7 h | 2.5 | H3PO2 (1 eq.), NaH2PO2 (3 eq.) | 2-MeTHF and water | 4e |
| 5 | Pd-polymer | 2 h | 0.2 | H2-1 atm, KOH (0.1 mol L−1) | Ethanol | 6c |
| 6 | Si-supported Pt | 0.5–2 h | 0.5–1.0 | H2-baloon | Methanol | 6d |
| 7 | Co–Co2Bf | 10–50 min | g | NaBH4 (4 eq.)/N2H4 (0.25 mL mmol−1) | Methanol–water | 11 |
| 8 | Ni–PAMAM–PVAm/SBA-15h | 2 h | — | NaBH4 (3 equiv.) | Water | 12 |
| 9 | Pd(OAc)2 | 30 min | 5 | PMHSi (3–5 eq.), KF (2 eq.) | THF | 7b |
| 10 | PVDF–[C6(mpy)2][NiCl4]22j | <15 min | 5 | NaBH4 (8 eq.) | Water | 9 |
| 11 | Au/TiO2 NPs | 30 min | 0.1 | NH3BH3 (1.5 eq.) | Ethanol | 13 |
| 12 | Zn-dust | 1–4 h | k | NH4Cl (2 eq.), TPGS-750 Ml | Water | 8 |
The reaction between a pyrazole derivatized hydroquinone ligand H2L and PdII(MeCN)2Cl2 in acetonitrile under reflux condition led to formation of complex 1 which was further purified by crystallization from acetonitrile solution (Scheme 1). The single crystal X-ray diffraction showed the complex crystallizes in orthorhombic, space group Cmc2(1). The N atoms from the two pyrazoles chelate to the PdII forming a seven membered ring and the remaining two coordination sites of the PdII are satisfied by two chloride ions. The two Pd–N bond distances are ca. 2.024 Å with an angle of ca. 85.5°. 1H NMR of the complex indicate that the complex is pure and supports that the solution structure is same as in the solid state where the Pd is coordinated via the two pyrazole nitrogen and the hydrogens of the two –OH groups of hydroquinone are visible in the 1H NMR spectra.
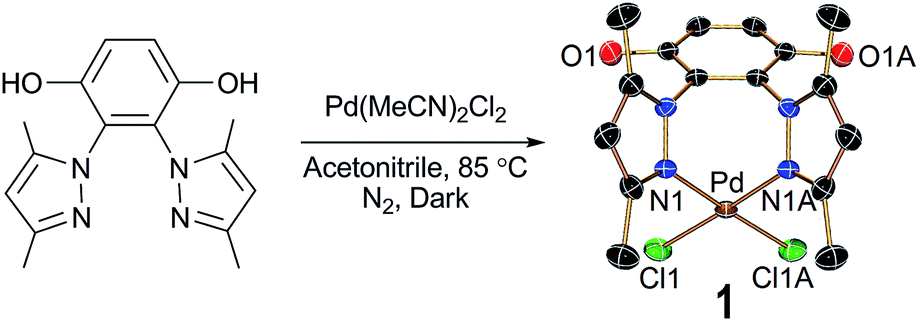 | ||
| Scheme 1 Synthetic methodology of 1 along with X-ray crystal structure displaying 30% probability thermal ellipsoids. All hydrogen atoms and solvent molecules are omitted for the sake of clarity. | ||
The catalytic activity studies of 1 towards the reduction of aromatic nitro compounds showed the catalyst to be a promising candidate. After optimization (Tables S3–S5, ESI†) it was found that with 0.25 mol% of catalyst at room temperature in water would provide with excellent isolated yield. Hence we probed the aromatic nitro reduction reactions with various substrates (Table 2) containing different functional group and found good efficiency and selectivity towards both aromatic and hetero aromatic nitro compounds. Most of the nitro substrates used are barely soluble in water but as the reaction proceeds they become soluble once converted to the respective amine. The progress of the reduction of 4-nitrobenzonitrile reaction was monitored by 1H NMR study in D2O at 25 °C (Fig. S3, ESI†) with catalyst 1, where we found the increase in product concentration, with respect to 1,4-dioxane used as standard, with increase in time, using 0.25 mol% catalyst. The NMR spectra also show the appearance of two peaks at 7.15 and 7.67 ppm due to the formation of hydroxylamine as an intermediate, which vanishes with time (Fig. S3†). It should be noted that the nitroarene reduction conditions used may also generate nanoparticles of Pd in solution although quantitative conversions were obtained using stoichiometric NaBH4. Hence to understand the importance of the ligand we studied the catalysis with other most common palladium substrates viz. Pd(OAc)2, PdCl2 and Pd(MeCN)2Cl2 only without the ligand H2L keeping other reaction conditions same (Table S6, ESI†). If only Pd is responsible for catalysis we would obtain similar yields in the nitroarene reduction. Ethanol was used as solvent since the palladium complexes used were better soluble in ethanol. Hence, the comparison would be more accurate in ethanol and the nitroarene reduction is equally efficient in ethanol. We found that using upto 1 mol% of the above three compounds the nitroarene reduction yield was less than 40% in 6 h whereas for the same substrate the [Pd(H2L)(Cl)2] (1) gave complete conversion to aryl amine in ca. 10 min emphasizing the efficiency of 1.
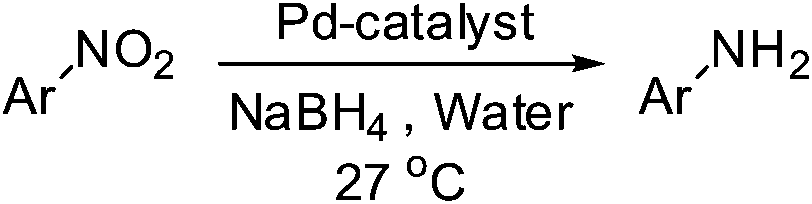
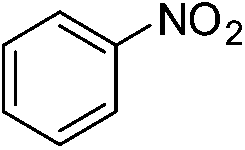
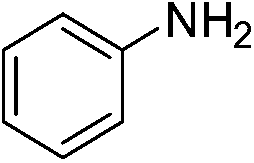
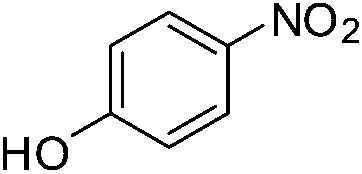
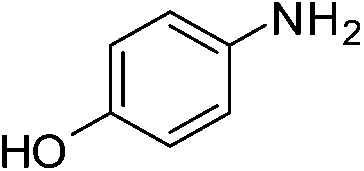
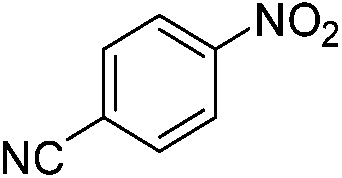
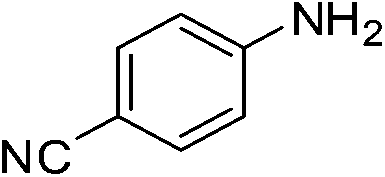

A careful look at Table 2 shows that with bromo or iodo substituted nitroarenes, the reduction reaction is coupled with dehalogenation. This indicated the probability of Suzuki–Miyaura cross-coupling by 1 where the dehalogenation is a part of the mechanistic step (Scheme S1, ESI†). The 1H NMR of the reaction between iodobenzene and NaBH4 in presence of 1 showed that the formation of a single peak at 7.32 ppm corresponding to the formation of benzene by dehalogenation of iodobenzene. There is also a weak signal at −17 ppm which might be due to the presence of a transient Pd–H intermediate14 (Fig. S1–S2, ESI†) indicating the role of Pd in dehalogenation during the reduction reaction. However, a direct hydride attack at the Pd bound benzene ring also cannot be excluded. The NMR shows that most of the substrate is converted to dehalogenated product by the time we recorded the NMR spectra (5–6 min) (Fig. S1, ESI†). Hence this catalyst has potential for dehalogenation and reduction in a single pot when the halogen atoms attached to the nitroarene is a bromine or iodine. A nitroarene reduction mechanism involving Pd–H intermediate may be as proposed in Scheme 2 (more details in Fig. S2, ESI†) which also takes into account the simultaneous dehalogenation. The mechanism is also supported by the literature knowledge of the transformation of quinone to hydroquinone in presence of hydride.15 In presence of sodium borohydride the quinone PdII complex 1 is reduced to a hydroquinone PdII complex which further changes to a quinone Pd0 complex and donates two electrons to the substrate/intermediate to perform two electron reduction in each step and the hydrogens are coming from solvent. After each step the quinone PdII complex 1 is reformed and the cycle continues. Hence, NaBH4 helps recycle the catalyst whereas the Pd centre donates electron to the substrate and consecutive intermediates helping the 6e− catalytic reduction (Schemes 2 and S2, ESI†). The chloro substituted substrates were not dehalogenated under similar conditions which reflect that it may be possible to impart selectivity of dehalogenation due to lower activity of the catalyst towards dehalogenation of chloro group. Since we found that the catalyst has possibility in Suzuki–Miyaura cross-coupling we probed it for the same as that would open a possibility of tandem catalysis using the same catalyst. The literature data suggested that a catalyst that would perform both Suzuki–Miyaura (with aryl halides and phenylboronic acids) and reduction of nitroarenes in a single pot in two consecutive steps in room temperature is not known.
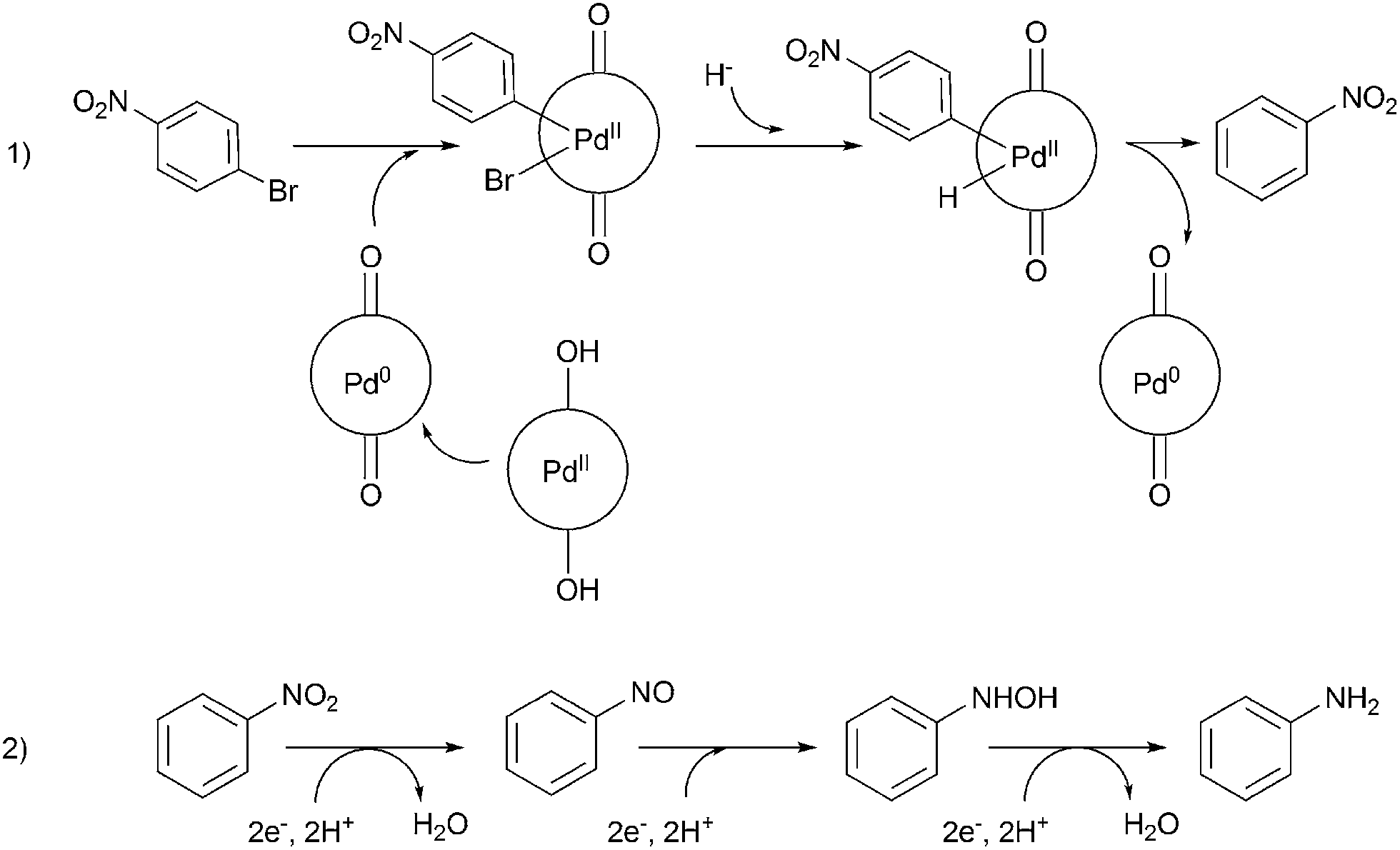 | ||
| Scheme 2 Representative dehalogenation and nitroarene reduction mechanism of 1-bromo-4-nitrobenzene as a model substrate (detailed mechanism in ESI†). | ||
The study of catalytic activity of 1 towards Suzuki–Miyaura cross coupling reaction was initially done with 4-bromoanisole which showed complete conversion with 0.5 mol% of catalyst. Following which the conditions were standardised varying the solvent (Table S7, ESI†), base (Table S8, ESI†) and catalyst loading (Table S9, ESI†) using the same halide substrate and phenylboronic acid. We found that ethanol and K2CO3 are the best solvent and base and only 0.25 mol% catalyst loading can successfully convert reactants to product within 1–4.5 h at room temperature using a green solvent like ethanol (with other substrates, Table S11, ESI†). It is well known that the palladium has to reduce to the Pd0 state to perform the catalytic cycle of Suzuki–Miyaura cross-coupling, here the hydroquinone ligand would be useful since the it is capable of undergoing two electron oxidation. Hence, the mechanistic steps proposed take this into account (Scheme S1, ESI†).
We attempted one pot tandem type catalysis to synthesize biaryl amines from nitro substituted aryl halide using 1. 0.25 mol% of 1 was loaded at the beginning of the C–C bond coupling reaction along with an aryl halide and phenylboronic acid in presence of K2CO3. Following the completion of C–C bond coupling monitored by silica gel TLC, NaBH4 was added as hydrogen source without adding any more catalyst and the biaryl amines were isolated in more than 97% yield of the desired products (Table 3).
|
|||
|---|---|---|---|
| R | X | Product | Isolated yieldb (%) |
| a Reaction condition: 1.0 mmol aryl nitro halide, 1.5 mmol phenylboronic acid, 3 mmol K2CO3, 0.25 mol% catalyst 1, ethanol at 27 °C; Followed by addition of 4 mmol NaBH4 in the same pot, 27 °C.b Isolated yields were reported after column chromatography. | |||
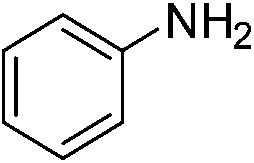
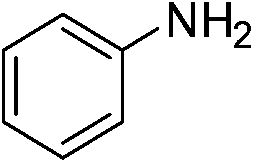
| p-NO2 | I | 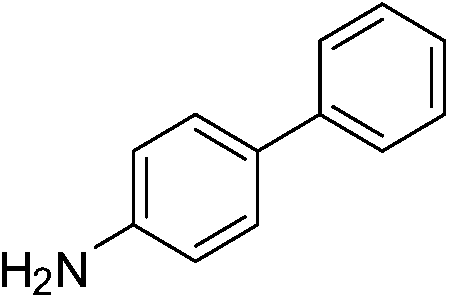 |
98 |
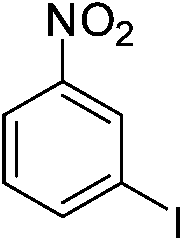
| m-NO2 | I | 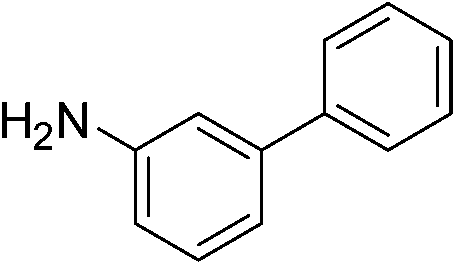 |
98 |
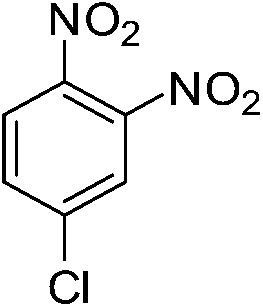
| p-NO2 | Br | 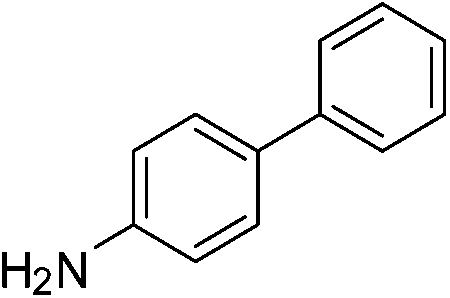 |
97 |
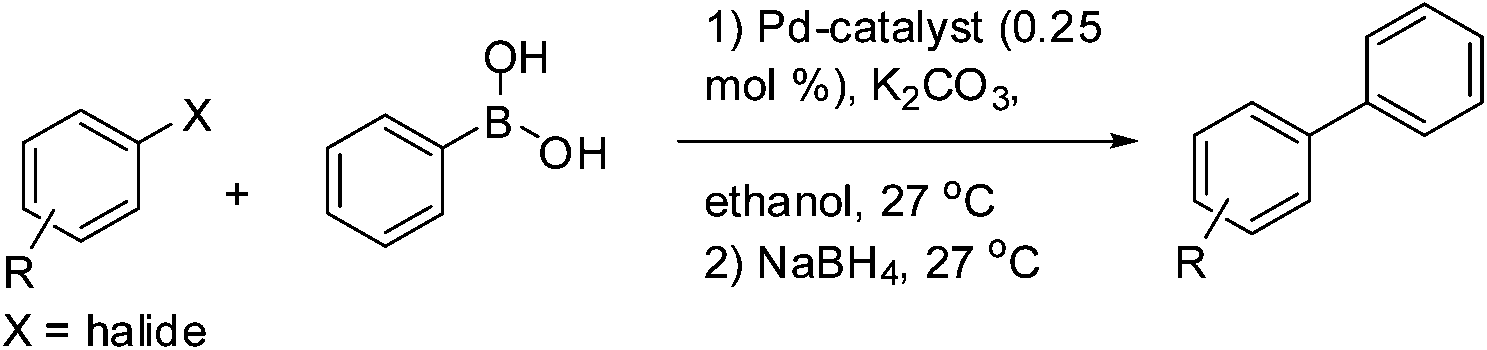
Conclusions
In conclusions, we have presented well characterized, easy to synthesize air stable hydroquinone based palladium complex 1 which shows promising activity towards reduction of nitroarenes in presence of other functionalities like nitriles. The amount of catalyst required is low (0.25 mol%) and moreover the same catalyst is also capable of C–C cross coupling followed by reduction of nitroarenes in one pot without any further catalyst addition which renders it suitable as a Tandem catalyst for the above purpose. Finally, for halide substrates the catalyst may have potential for simultaneous dehalogenation and nitro reduction if the halides are bromine or iodine. The potential of the catalyst warrants further work with a wider spectrum of substrate to check upon its functionality tolerance and use of the dehalogenation reaction for other possible substitutions along with detailed mechanistic studies.Acknowledgements
We sincerely acknowledge DST for financial support vide project no SR/S1/IC-36/2010. We also thank IISER Kolkata for the financial and infra-structural support, including NMR, single crystal X-ray facilities and other analytical instruments. A.K. is thankful to DST for Inspire fellowship. K.P. thanks UGC, A.S. thanks IISER Kolkata and S.K.D. thanks CSIR-India for providing research fellowship.Notes and references
- (a) M. K. Lakshman, J. Organomet. Chem., 2002, 653, 234 CrossRef CAS; (b) A. Suzuki, J. Organomet. Chem., 1999, 576, 147 CrossRef CAS; (c) U. Christmann and R. Vilar, Angew. Chem., Int. Ed., 2005, 44, 366 CrossRef CAS PubMed; (d) S.-D. Yang, C.-L. Sun, Z. Fang, B.-J. Li, Y.-Z. Li and Z.-J. Shi, Angew. Chem., Int. Ed., 2008, 47, 1473 CrossRef CAS PubMed; (e) A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722 CrossRef CAS PubMed; (f) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Commun., 2010, 46, 677 RSC; (g) F. K. Sheffy, J. P. Godschalx and J. K. Stille, J. Am. Chem. Soc., 1984, 106, 4833 CrossRef CAS; (h) R. Giri, N. Maugel, J.-J. Li, D.-H. Wang, S. P. Breazzano, L. B. Saunders and J.-Q. Yu, J. Am. Chem. Soc., 2007, 129, 3510 CrossRef CAS PubMed; (i) K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2007, 129, 11904 CrossRef CAS PubMed; (j) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS; (k) A. F. Littke and G. C. Fu, Angew. Chem., Int. Ed., 2002, 41, 4176 CrossRef CAS; (l) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CAS; (m) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442 CrossRef CAS PubMed; (n) A. F. Littke, C. Dai and G. C. Fu, J. Am. Chem. Soc., 2000, 122, 4020 CrossRef CAS.
- (a) D. Hollmann, S. Baehn, A. Tillack and M. Beller, Angew. Chem., Int. Ed., 2007, 46, 8291 CrossRef CAS PubMed; (b) S. U. Sonavane, M. B. Gawande, S. S. Deshpande, A. Venkataraman and R. V. Jayaram, Catal. Commun., 2007, 8, 1803 CrossRef CAS PubMed; (c) X. Yuan, N. Yan, C. Xiao, C. Li, Z. Fei, Z. Cai, Y. Kou and P. J. Dyson, Green Chem., 2010, 12, 228 RSC; (d) F. Wang, J. Liu and X. Xu, Chem. Commun., 2008, 2040 RSC; (e) A. Yokoyama, H. Suzuki, Y. Kubota, K. Ohuchi, H. Higashimura and T. Yokozawa, J. Am. Chem. Soc., 2007, 129, 7236 CrossRef CAS PubMed; (f) K. C. Nicolaou, J. M. Ramanjulu, S. Natarajan, S. Brase, H. Li, C. N. C. Boddy and F. Rubsam, Chem. Commun., 1997, 1899 RSC; (g) O. Baudoin, M. Cesario, D. Guenard and F. Gueritte, J. Org. Chem., 2002, 67, 1199 CrossRef CAS PubMed.
- (a) R. S. Downing, P. J. Kunkeler and H. van Bekkum, Catal. Today, 1997, 37, 121 CrossRef CAS; (b) A. M. Tafesh and J. Weiguny, Chem. Rev., 1996, 96, 2035 CrossRef CAS PubMed; (c) R. J. Rahaim, Jr and R. E. Maleczka, Jr, Org. Lett., 2005, 7, 5087 CrossRef PubMed; (d) S. Chandrasekhar, S. J. Prakash and C. L. Rao, J. Org. Chem., 2006, 71, 2196 CrossRef CAS PubMed; (e) B. Chen, U. Dingerdissen, J. G. E. Krauter, H. G. J. Lansink Rotgerink, K. Moebus, D. J. Ostgard, P. Panster, T. H. Riermeier, S. Seebald, T. Tacke and H. Trauthwein, Appl. Catal., A, 2005, 280, 17 CrossRef CAS PubMed; (f) P. K. Mandal and J. S. McMurray, J. Org. Chem., 2007, 72, 6599 CrossRef CAS PubMed.
- (a) R. Dey, N. Mukherjee, S. Ahammed and B. C. Ranu, Chem. Commun., 2012, 48, 7982 RSC; (b) S. Park, I. S. Lee and J. Park, Org. Biomol. Chem., 2013, 11, 395 Search PubMed; (c) X. Liu, S. Ye, H.-Q. Li, Y.-M. Liu, Y. Cao and K.-N. Fan, Catal. Sci. Technol., 2013, 3, 3200 RSC; (d) K. Layek, M. L. Kantam, M. Shirai, D. Nishio-Hamane, T. Sasaki and H. Maheswaran, Green Chem., 2012, 14, 3164 RSC; (e) M. Baron, E. Metay, M. Lemaire and F. Popowycz, Green Chem., 2013, 15, 1006 RSC; (f) A. K. Shil and P. Das, Green Chem., 2013, 15, 3421 RSC.
- (a) I. H. Abd El Maksod, E. Z. Hegazy, S. H. Kenawy and T. S. Saleh, Adv. Synth. Catal., 2010, 352, 1169 CrossRef CAS PubMed; (b) R. V. Jagadeesh, G. Wienhoefer, F. A. Westerhaus, A.-E. Surkus, M.-M. Pohl, H. Junge, K. Junge and M. Beller, Chem. Commun., 2011, 47, 10972 RSC; (c) P. P. Sarmah and D. K. Dutta, Green Chem., 2012, 14, 1086 RSC; (d) T. Schabel, C. Belger and B. Plietker, Org. Lett., 2013, 15, 2858 CrossRef CAS PubMed; (e) M. Sutter, L. Pehlivan, R. Lafon, W. Dayoub, Y. Raoul, E. Metay and M. Lemaire, Green Chem., 2013, 15, 3020 RSC; (f) F. Yuste, M. Saldana and F. Walls, Tetrahedron Lett., 1982, 23, 147 CrossRef CAS; (g) K. Junge, B. Wendt, N. Shaikh and M. Beller, Chem. Commun., 2010, 46, 1769 RSC; (h) U. Sharma, P. K. Verma, N. Kumar, V. Kumar, M. Bala and B. Singh, Chem.–Eur. J., 2011, 17, 5903 Search PubMed.
- (a) G. Brieger and T. J. Nestrick, Chem. Rev., 1974, 74, 567 CrossRef CAS; (b) P. Lara, A. Suarez, V. Colliere, K. Philippot and B. Chaudret, ChemCatChem, 2014, 6, 87 CrossRef CAS PubMed; (c) X. Xi, Y. Liu, J. Shi and S. Cao, J. Mol. Catal. A: Chem., 2003, 192, 1 CrossRef CAS; (d) V. Pandarus, R. Ciriminna, F. Beland and M. Pagliaro, Adv. Synth. Catal., 2011, 353, 1306 CrossRef CAS PubMed; (e) R. F. D'Vries, M. Iglesias, N. Snejko, S. Alvarez-Garcia, E. Gutierrez-Puebla and M. A. Monge, J. Mater. Chem., 2012, 22, 1191 RSC.
- (a) L. Pehlivan, E. Metay, S. Laval, W. Dayoub, P. Demonchaux, G. Mignani and M. Lemaire, Tetrahedron Lett., 2010, 51, 1939 Search PubMed; (b) R. J. Rahaim, Jr and R. E. Maleczka, Jr, Synthesis, 2006, 3316 Search PubMed.
- S. M. Kelly and B. H. Lipshutz, Org. Lett., 2014, 16, 98 Search PubMed.
- A. Chinnappan and H. Kim, RSC Adv., 2013, 3, 3399 Search PubMed.
- (a) P. Anastas and J. Warner, Green Chemistry: Theory and Practice, 1998 Search PubMed; (b) J. Clark and D. MacQuarrie, Handbook of Green Chemistry & Technology, 2002 Search PubMed; (c) M. Lancaster, Green Chemistry: An Introductory Text, 2002 Search PubMed.
- A. A. Vernekar, S. Patil, C. Bhat and S. G. Tilve, RSC Adv., 2013, 3, 13243 RSC.
- R. J. Kalbasi and F. Zamani, RSC Adv., 2014, 4, 7444 RSC.
- E. Vasilikogiannaki, C. Gryparis, V. Kotzabasaki, I. N. Lykakis and M. Stratakis, Adv. Synth. Catal., 2013, 355, 907 CrossRef CAS PubMed.
- (a) P. Leoni, M. Sommovigo, M. Pasquali, S. Midollini, D. Braga and P. Sabatino, Organometallics, 1991, 10, 1038 CrossRef CAS; (b) Y. Zhu, C.-H. Chen, C. M. Fafard, B. M. Foxman and O. V. Ozerov, Inorg. Chem., 2011, 50, 7980 CrossRef CAS PubMed.
- (a) J. Cheng, K. L. Handoo, J. Xue and V. D. Parker, J. Org. Chem., 1993, 58, 5050 CrossRef CAS; (b) X.-Q. Zhu, C.-H. Wang, H. Liang and J.-P. Cheng, J. Org. Chem., 2007, 72, 945 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: General synthesis procedure of ligand and metal complex, characterization data, single crystal X-ray data of 1, NMR spectra, complete optimization data of catalytic reaction, standard procedure of catalysis, mechanism of catalytic cycle, table of Suzuki–Miyaura cross coupling reaction. CCDC 996012. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra06547f |
| ‡ The authors pay equal contributions to this work. |
| This journal is © The Royal Society of Chemistry 2014 |