
Synthetic approach to novel azido esters and their utility as energetic plasticizers†
Dimple Kumaria,
Anjitha S. G.b,
Chandra Shekhar Pantc,
Mahendra Patilb,
Haridwar Singha and
Shaibal Banerjee*a
aDepartment of Applied Chemistry, Defence Institute of Advanced Technology (DU), Girinagar, Pune 411025, India. E-mail: shaibal.b2001@gmail.com; Fax: + 91 20 2438 9411; Tel: + 91 20 2430 4164
bCentre for Excellence in Basic Sciences, University of Mumbai, Vidhyanagari Campus, Mumbai 400098, India
cAdvanced Centre for Energetic Materials, DRDO, Nasik 422207, India
First published on 13th August 2014
Abstract
A modular approach towards a series of novel di azido, tetra azido and hexa azido esters is introduced here. The methodologies adopted towards their synthesis are short, environmentally friendly, cheap and scalable. Detailed computational, physical and thermal studies of these plasticizers with binders have been carried out. Our results reveal impressive properties in terms of thermal stability, sensitivity, nitrogen content, oxygen balance, kinetics and heat of formation of synthesized plasticizers and their compatibility with energetic binders.
Introduction
With the discovery of organic azides by Peter Griess, numerous syntheses of energy-rich molecules have been developed.1,2 As the azido group is a highly energetic functional group, compounds containing azido groups often exhibit explosive properties. The N3 π-bond can easily be polarized due to lone pair repulsion which consequently results in strong exothermic dissociation reactions with the release of molecular nitrogen and reactive nitrene groups. In general, the introduction of an azido group into an organic compound increases its energy content by approx. 290–355 kJ mol−1.3,4Due to this, organic azides are considered and partly used as energetic materials such as energetic polymers or high energy density materials (HEDM) in propellant formulations.5–7
Compounds of this category include azido polyethers, azido nitramines, azido nitro compounds, azido alkanes and azido esters.8–15 Organic azides also use as precursor for synthesis of various heterocyclic compounds like triazoles, tetrazoles etc. and thus commercial interest of organic azide compounds are worth pursuing.16–22
Azido esters are used as energetic plasticizers which hold tremendous promise for propellants formulation and envisaged to replace conventional plasticizers in near future.23,24 Usually majority of plasticizers have ester group within them since ester functionality is known to have lubricating effect. Plasticizers help in improving the mechanical properties of the propellant grain. These function by positioning themselves in between the polymeric binder's chain to reduce chain–chain interaction i.e. decrease the intermolecular forces between the polymer chains, which results in the softening polymeric composition. They increase polymer's elongation capacity and processability by lowering the glass transition temperature and viscosity of the binder-plasticizer blends.25 The basic objectives of developing energetic plasticizers are to enhance the thermal stability, energy content, oxygen balance and burning behaviour of the formulation.26–30 By taking account of all these properties, together with better stability and good compatibility with azido polymeric binders like glycidyl azide polymer (GAP), bis azidomethyl oxetane (BAMO) etc. as compared to nitrate ester plasticizers like trimethylolethane trinitrate (TMETN) and 1, 2, 4-butanetriol trinitrate (BTTN) which improves processibility and solid loading of azido binders azido esters have received increasing attentions in recent time.
Various multi azido-ester plasticizers are like- bis(2-azidoethyl)adipate (BAEA), ethylene glycol bis azido acetate (EGBAA), diethylene glycol bis azido acetate (DEGBAA), trimethylol nitro methane triazido acetate (TMNTA), pentaerythritol tetrakis azido acetate (PETKAA), bis azido diethyl phthalate (BADEP), bis azido dipropyl phthalate (BADPP), bis azido dibutyl phthalate (BADBP), tris(azido acetoxy methyl)propane (TAAMP), bis (azido acetoxy) bis(azido methyl)propane (BABAMP), di(azido acetoxy) di(azido methyl) propane (PEAA), bis(1,3-diazido prop-2-yl)malonate, bis(1,3-diazido prop-2-yl)glutarate, and dendritic azido esters have been reported.31–38 All these reported azido esters, except dendrimers, are synthesized along with similar route of ester formation by reflux reaction followed by substitution of halogen with azide anion (shown in Scheme 1). Usually esterification process involves mixing of acid, alcohol and catalytic amount of sulphuric acid or p-toluene sulfonic acid and refluxed. The second step is azidation of chloro ester, obtained from first step; get corresponding azides which is nucleophilic bimolecular substitution reaction (SN2).
 | ||
| Scheme 1 Synthesis of azido ester. | ||
In case of dendritic azido ester, during esterification Pant et al. have used dimethyl amino pyridinium-p-toluenesulfonate (DPTS) and N,N-dicyclohexylcarbodiimide (DCC) at 25 °C.33
Our group is involved in the synthesis of tetra and hexa azido esters. We have used modular approach i.e. synthesis of various di, tetra and hexa azido esters from a common precursor. The methodology adopted again involves two step syntheses, the first step is azidation of alcohol followed by esterification of azido alcohol with carboxylic acids to get corresponding azido ester (6a–6m) with the use of coupling reagents N,N′- dicyclohexylcarbodiimide (DCC) and 4-(dimethylamino)pyridine (DMAP) at room temperature which is also favourable from safety point of view. These reactions are represented as Schemes 2 & 3.
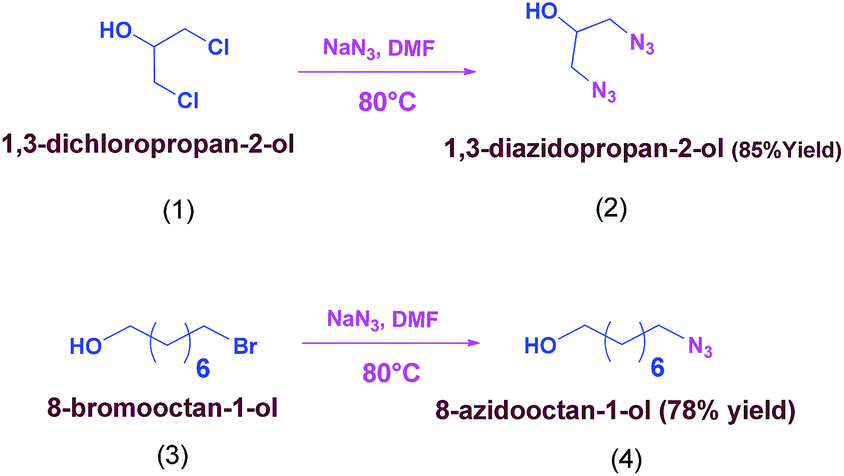 | ||
| Scheme 2 Synthesis of azido alcohols. | ||
 | ||
| Scheme 3 Synthesis of Azido esters. | ||
We have taken 1,3-dichloropropane-2-ol (1) and 8-bromooctan-1-ol (3) as halogen containing alcohol and 3,5-dinitro benzoic acid, 3-trifluoro methyl-4-nitro benzoic acid, 4-nitro-3-carboxylic pyrazole, 4-nitro phthalic acid, homo phthalic acid, indane dicarboxylic acid, cyclobutane-1,1-dicarboxylic acid, 3,5-pyridinedicarboxylic acid, tricarballylic acid, nitrilotriacetic acid and benzene-1,2,3- tricarboxylic acid as carboxylic acids to get various novel azido esters shown in Fig. 1.
 | ||
| Fig. 1 Novel di, tetra and hexa azido esters. | ||
Aromatic moiety exhibits thermal stability. Dioctyl phthalate (DOP) is the most widely used inert plasticizer because of its stability and good lubricating effect, so we have tried to make it energetic by embedding –N3 and –NO2 groups as (6d) and (6e). Similarly we have synthesized –CF3 containing azido ester (6b) which increases the thermal and chemical stability and the density of organic molecule. Strain ring (6h) and bicycle (6f) azido esters are known to give the good combinations of stability and large positive heats of formation. However there are no reports on incorporation of strained moiety in energetic plasticizers. This motivated us to synthesize azido ester having strained cyclobutane ring. Heterocycles generally have a higher heat of formation, density, and oxygen balance than their carbocyclic analogues. Also the lone pair on nitrogen is known to coordinate with Lewis acids like metals. Therefore we have targeted (6c) and (6g). Hexa azido esters posses large challenges since N/C ratio is quite high and hence have not been reported in the literature presumably due to the instability associated with several azide groups at close proximity. So as an extrapolation, we ventured to synthesis hexa azido esters. By following the procedure in Schemes 1 and 2, we were able to synthesize hexa azido esters (6k), (6l) and (6m) in good yield. These classes of plasticizers will be a niche of its kind. Representative spectrum of 15N NMR of (6l) is depicted below in Fig. 2.
 | ||
| Fig. 2 15N NMR spectrum of tris(1,3-diazidopropan-2-yl) −2,2′,2′-nitrilotriacetate (6l). | ||
Results and discussion
Computational studies
The heat of formation of newly designed azido-esters (6a to 6m) using density functional methods (DFT) methods. All calculations were performed using Gaussian 09 suite of programmes.39 Several chemically intuitive guess structures, mainly by rotating different moieties attached to the side chains (–CO–O–CH(CH2N3)2), were considered for the structural optimization of azido-esters at the M062X/6-31G* level of theory.40 Only the lowest-energy conformers of azido-esters are reported in this paper. All structures were identified as minima using harmonic force constant analysis at the same level of theory. The heat of formation (ΔfH) is a crucial parameter in theoretical studies of energetic materials.41 Isodesmic reactions shown in the Schemes 4–6 were considered to calculate the reaction enthalpy (ΔHRXN) at 298 K. The heats of formation (ΔfH°) of azido-esters listed in the Table 1 were determined from the computed ΔHRXN values and the known ΔfH° data of the other reactants and products (see ESI for details†). In order to compare the heat of formation, we have grouped azido-esters into three schemes; Scheme 4 contains azido-ester having phenyl ring (6a, 6b, 6i, 6j and 6m), Scheme 5 includes azido-esters having heterocyclic ring (6c and 6g) and Scheme 6 contains azido-esters without any aromatic ring (6k and 6l). The heat of formation of other azido-esters could not be estimated since experimental data for heats of formation of the reference molecules in the isodesmic reactions was not available in the literature. | ||
| Scheme 4 Isodesmic reactions for the calculation of heat of formation of 6a, 6b, 6i, 6j and 6m. | ||
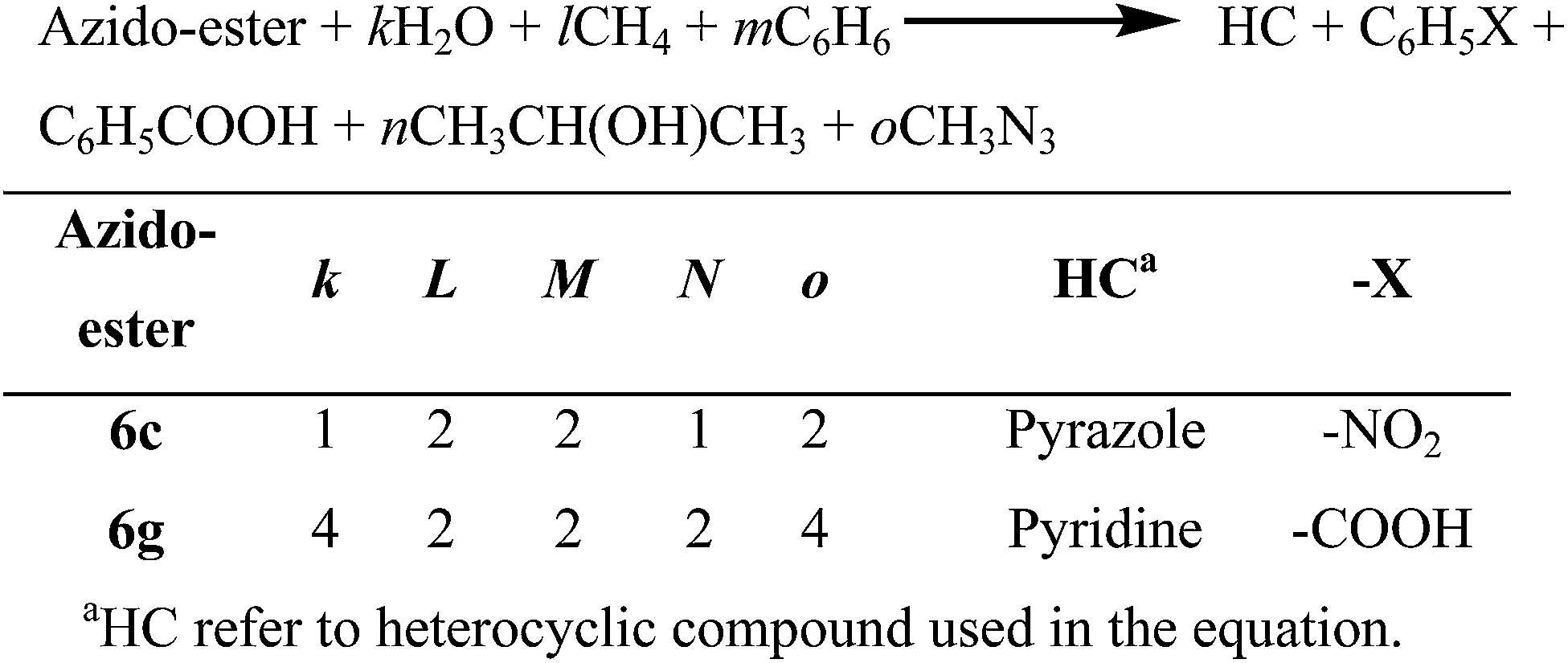 | ||
| Scheme 5 Isodesmic reactions for the calculation of heat of formation of 6c and 6g. | ||
 | ||
| Scheme 6 Isodesmic reactions for the calculation of heat of formation of 6k and 6l. | ||
| Azido ester | ΔfH° kJ mol−1 | Azido ester | ΔfH° kJ mol−1 | Azido ester | ΔfH° kJ mol−1 |
|---|---|---|---|---|---|
| 6a | 251.6 | 6c | 363.3 | 6k | 827.6 |
| 6b | 344.3 | 6j | 650.3 | 6l | 1422.6 |
| 6h | 547.3 | ||||
| 6i | 766.5 | ||||
| 6m | 791.1 |
The values of heat of formations of azido-esters 6a, 6b, 6i, 6j and 6m as shown in Table 1 have consistently increased with the increasing number of azido-ester group attached to the phenyl ring (ΔfH°: 6a<6i < 6m). With each substitution of azido-ester group (–COOCH(CH2N3)2) to the phenyl ring, heat of formation is found to be increased by 200–250 kJ mol−1. This trend is fairly similar to energetic materials containing azide group in which substitution of azide group increases energy content of molecule by 290–330 kJ mol−1.42 In this series of azido-esters, 6m with six azido-ester groups have the highest heat of formation (ΔfH°: 791.1 kJ mol−1) whereas di-azido-ester 6a possess the lowest heat of formation (ΔfH°: 251.6 kJ mol−1). To verify substituents effect (–NO2 and CF3) in the case of azido-esters 6a and 6b, the heat of formation of benzene substituted azido ester (–COOCH(CH2N3)2) is also calculated via isodesmic reaction. The calculated heat of formation for the benzene substituted azido-ester is 390.5 kJ mol−1 which is higher than azido ester 6a and 6b. Similar trend in ΔfH° of azido-esters 6i and 6j is noticed where azido-ester without –NO2 substitution on benzene ring 6j (ΔfH°: 888.1 kJ mol−1) possess higher heat of formation than azido-ester without –NO2 substitution on benzene ring 6i (ΔfH°: 645.3 kJ mol−1). These results suggest –NO2 and –CF3 substitution on the benzene ring may not increase the heat of formation of azido-ester.
The heat of formation of azido-esters containing heterocyclic ring (6c and 6g) were calculated via isodesmic reactions shown in the Scheme 5. While the pyrazole containing azido-ester 6c (ΔfH°:363.3 kJ mol−1) have the heat of formation comparable to other di-azido-esters (6a and 6b), the pyridine containing azido-ester 6g (ΔfH°:650.0 kJ mol−1) possess a fairly high heat of formation compared to tetra-azido ester (6i). Next, the heats of formation of azido-esters without aromatic ring (6k and 6l) (Scheme 6) are compared. These azido-esters have substantially high heat of formation compared to the azido-esters containing aromatic ring (6m). Especially, azido-esters 6l possess the heat of formation of about 1422.6 kJ mol−1, which is the highest ΔfH° value among the all azido-esters.
In order to verify thermal stability of the azido-esters, we have examined bond orders (BO) and bond dissociation energies (BDE) of selected bonds of azido-esters (6a). The bond distances, bond orders and bond dissociation energies of selected bonds are given in the Table 2. The calculated BDEs of bonds of side-chain of azido-esters are higher than 200 kJ mol−1. This indicates that azido-esters are thermally stable molecules and rather high temperatures may be required for breaking bonds of side-chain of azido-esters. However, at elevated temperature, bond cleavage may start at N1–N2 or C4– N1 bond since these bonds have the lowest BDE, 238.4 and 349.9 kJ mol−1, respectively.Fig. 3
| Bond distance | Bond order | Bond dissociation energy (BDE) | |
|---|---|---|---|
| C1–C2 | 1.49 | 0.89 | 491.4 |
| C2–O2 | 1.34 | 0.89 | 480.6 |
| C3–O2 | 1.43 | 0.68 | 427.3 |
| C3–C4 | 1.44 | 0.83 | 477.2 |
| C4–N1 | 1.47 | 0.79 | 379.9 |
| N1–N2 | 1.23 | 1.77 | 238.3 |
| N2–N3 | 1.13 | 1.63 | 734.8 |
 | ||
| Fig. 3 Optimized structure of (6a) (numbers are given to selected atoms). | ||
Thermal studies by DSC and TGA and Sensitivity studies.
DSC experiments showed high thermal stability of all azido esters over a wide temperature range (up to 200 °C). Thus all compounds are nonhazardous in nature under ambient conditions. The decomposition enthalpy or heat release is remarkable as compared to other reported azido ester plasticizers.The weight loss pattern of all compounds is depicted in their respective TGA thermo gram and it was found to be similar in some way. All compounds showed stepwise decompositions. At the first step, all azido groups decomposed to produce nitrogen and at the second step, ester groups (–CH2OCO–) followed by carbon backbone were found to be decomposed.36 Generally, organic azide decomposes stepwise during slow heating rate. However, the main exothermicity of the thermal decomposition is related only to the first mass loss step which corresponds well with the release of molecular nitrogen as a result of azide decomposition.
The glass transition temperature (Tg) value for all synthesized liquid azido esters are in the range of −75.97 °C to −20.84 °C. The low Tg value of these compounds as well as reported azido esters makes them ideal candidates as plasticizers for use in propellants formulations.34–38
Sensitivity of energetic materials for impact, friction are also important parameters to observe and are mandatory for assessing hazard in manufacture, handling, storage and transportation. The susceptibility of these stimuli, results in either combustion and detonation. For all azido esters, impact sensitivity test results showed that all compounds were insensitive to mechanical stimuli up to 33.32 Nm. Friction sensitivity test results showed that all compounds were insensitive up to 352.8 N.
All synthesized azido esters have ‘negative’ OB (%) in the range of −76.86 to −213.99 which are comparable with many reported energetic plasticizers except nitroglycerine (+03.50%). The nitrogen content of all thirteen compounds are found to be in the range of 17.27% to 47.20% out of which six azido esters having more than 40% nitrogen content which is fairly same as the reported value for high nitrogen content energetic plasticizers.34–38 (N2 content, OB, decomposition temperature, heat release, Tmax and TGA values of synthesized azido esters are given in Table 3).
| Azido esters | NC (%) | OB | DT (°C) | H r (J g−1) | Tmax (°C) | TGA |
|---|---|---|---|---|---|---|
| (6a) | 33.31 | −85.71 | 249.63 | 2362 | 230.12 | 52.17% 200–280 °C |
| (6b) | 27.28 | −98.05 | 250.83 | 2113 | 225.24 | 85.26% 125–305 °C |
| (6c) | 44.81 | −76.86 | 249.43 | 2098 | 230.16 | 55.27% 180–304 °C |
| (6d) | 18.93 | −184.13 | 268.33 | 1819 | 265.48 | 22.52% 200–280 °C |
| (6e) | 17.27 | −213.99 | 268.83 | 1018 | 244.24 | 4.92% 140–195 °C 16.32% 195–225 °C 15.99% 225–310 °C |
| (6f) | 36.97 | −137.32 | 233.50 | 1614 | 262.18 | 90.11% 160–275 °C |
| (6g) | 43.82 | −109.87 | 249.56 | 1886 | 249.56 | 42.23% 180–290 °C |
| (6h) | 42.82 | −114.29 | 243.04 | 1407 | 243.04 | 83.53% 180–270 °C |
| (6i) | 39.62 | −99.27 | 209.83 | 1964 | 244.98 | 66.02% 180–280 °C |
| (6j) | 39.22 | −126.99 | 237.23 | 1565 | 229.12 | 47.24% 190–265 °C |
| (6k) | 45.94 | −84.67 | 242.48 | 1602 | 242.48 | 48.25% 200–280 °C |
| (6l) | 47.20 | −107.22 | 242.34 | 1876 | 242.34 | 53.62% 180–280 °C 18.60% 115–205 °C 28.20% |
| (6m) | 43.27 | −107.22 | 246.38 | 2244 | 242.48 | 18.60% 115–205 °C 28.20% 205–280 °C 25.37% 280–420 °C |
Thermal decomposition kinetics of 6c, 6g & 6l
A good understanding of reaction kinetics of exothermic decomposition of the azido compounds can be obtained by Differential Scanning Calorimetry (DSC).43–47The Arrhenius parameters viz. activation energy (E) and pre-exponential factor (A) for the exothermic decomposition of 1,3-diazidopropan-2-yl 4-nitro- 1H-pyrazole-3-carboxylate (6c), bis(1,3-diazidopropan-2-yl)pyridine-3,5-dicarboxylate (6g) and tris(1,3-diazidopropan-2-yl) 2,2′,2′′-nitrilotriacetate (6l) were obtained using non-isothermal Kissinger method.48–50 This method allows to obtain the value of E and A from a plot of ln(β/T2) against 1000/T for a series of experiments at different heating rates (β), where T is the peak temperature of the exothermic decomposition in DSC thermogram. The equation is as follows
 | (1) |
Straight lines obtained from the plot of ln(β/T2) against 1000/T (Fig. 4), which indicated that the mechanism of thermal decomposition of these compounds did not vary during the decomposition under various heating rates. The activation energy and pre-exponential factor were derived from the slope and intercept of plotting regression line, respectively. In order to calculate the pre-exponential factor, it was assumed that the decomposition of these compounds followed first-order kinetics.
 | ||
| Fig. 4 The plot of ln(β/T2) versus 1000/T for 6c, 6j & 6l. | ||
The results obtained for activation energy and pre-exponential factor are listed in Table 4. The activation energies for three azido esters are similar to that of reported azido compounds i.e. in the range of 164–172 kJ mol−1.25 This result is conclusive evidence to the fact that azido compounds, irrespective of their difference in structure, decompose primarily by the scission of the azide bond.
| Compounds | E (kJ mol−1) | ln A (min−1) |
|---|---|---|
| (6c) or (DK-06-271) | 105.43 | 23.85 |
| (6g) or (DK-06-212) | 150.21 | 34.24 |
| (6l) or (DK-06-243) | 178.52 | 41.41 |
Compatibility with energetic binders
To study the compatibility of azido esters with energetic binders like GAP and BAMO, all synthesized viscous liquid azido esters (except three azido esters which are solids i.e. 6a, 6c and 6f) were mixed with these binders at a ratio of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :80 (w/w) thoroughly at room temperature and resultant mixtures were kept at room temperature to observe any phase separation. No layer separation or any other sign of heterogeneity was found. This homogeneity indicates the physical compatible of azido esters with both binders. Further, all these binder plasticizer combinations show reduction in glass transition temperatures. This is due to the reduction in cohesive forces of attraction between polymer chains. Plasticizers when added to polymer significantly reduce the brittleness by penetrating deep inside the polymer matrix and reducing the cohesive forces between polymers and increase the free volume. This causes an increase in segment mobility, leading to reduction of Tg.23–25 The single point Tg values for all combinations (shown in Table 5) another evidence of the presence of single phase homogeneous system which confirms the thermodynamic compatibility of azido esters with these energetic binders.34–38
:80 (w/w) thoroughly at room temperature and resultant mixtures were kept at room temperature to observe any phase separation. No layer separation or any other sign of heterogeneity was found. This homogeneity indicates the physical compatible of azido esters with both binders. Further, all these binder plasticizer combinations show reduction in glass transition temperatures. This is due to the reduction in cohesive forces of attraction between polymer chains. Plasticizers when added to polymer significantly reduce the brittleness by penetrating deep inside the polymer matrix and reducing the cohesive forces between polymers and increase the free volume. This causes an increase in segment mobility, leading to reduction of Tg.23–25 The single point Tg values for all combinations (shown in Table 5) another evidence of the presence of single phase homogeneous system which confirms the thermodynamic compatibility of azido esters with these energetic binders.34–38
| S. No. | Azido esters | Tg | S. No. | Azido esters | Tg |
|---|---|---|---|---|---|
| 1 | GAP | −49.78 | 18 | (6h) + GAP | −52.49 |
| 2 | BAMO | −53.77 | 19 | (6h) + BAMO | −56.09 |
| 3 | (6a) | Solid | 20 | (6i) | −34.38 |
| 4 | (6b) | −42.24 | 21 | (6i) + GAP | −53.90 |
| 5 | (6b) + GAP | −38.82 | 22 | (6i) + BAMO | −59.75 |
| 6 | (6b) + BAMO | −48.22 | 23 | (6j) | −55.48 |
| 7 | (6c) | Solid | 24 | (6j) + GAP | −50.11 |
| 8 | (6d) | −75.97 | 25 | (6j) + BAMO | −53.60 |
| 9 | (6d) + GAP | −53.90 | 26 | (6k) | −55.66 |
| 10 | (6d) + BAMO | −59.75 | 27 | (6k) + GAP | −51.30 |
| 11 | (6e) | −52.25 | 28 | (6k) + BAMO | −55.38 |
| 12 | (6e) + GAP | −50.11 | 29 | (6l) | −45.99 |
| 13 | (6e) + BAMO | −53.60 | 30 | (6l) + GAP | −49.47 |
| 14 | (6g) | −41.01 | 31 | (6l) + BAMO | −53.93 |
| 15 | (6g) + GAP | −50.25 | 32 | (6m) | −20.84 |
| 16 | (6g) + BAMO | −54.71 | 33 | (6m) + GAP | −52.98 |
| 17 | (6h) | −67.49 | 34 | (6m) + BAMO | −47.67 |
Experimental section
Materials
All chemicals used were obtained from commercial suppliers and were used without further purification. All the reactions were monitored by precoated silica gel 60 F254 (Merck) TLC plates.1H-NMR and 13C-NMR spectra were recorded on a Varian mercury-300 MHz and 75 MHz instrument, respectively using tetramethylsilane (TMS) as an internal reference and CDCl3 as solvent. 13C-NMR DEPT and 15N NMR spectra were recorded on a BRUKER- 100 MHz and 40 MHz instrument respectively using tetramethylsilane (TMS) as an internal reference with CDCl3 and DMSO as solvents respectively. 1H-COSY NMR and 19F-NMR were recorded on a BRUKER- 400 MHz and 374.96 MHz instrument respectively using tetramethylsilane (TMS) as an internal reference with CDCl3. Coupling constants (J values) are given in Hertz (Hz). Chemical shifts are expressed in parts per million (ppm) downfield from internal reference, tetramethylsilane. The standard abbreviations s, d, t, q, quin, m and dd refer to singlet, doublet, triplet, quartet, quintet, multiplet and doublet of doublet respectively. IR spectra were recorded on a BRUKER ALPHA ECO ATR FT-IR spectrometer as a neat sample in the range of 500 to 4000 cm−1. High resolution mass spectra measurements were carried out on Micromass Q-Tof Mass spectrometer. Melting points were recorded on BUCHI M560.
Differential Scanning Calorimetry (DSC) studies were carried out on a Perkin Elmer DSC-7 instrument operating at a heating rate of 10 °C min−1 in nitrogen atmosphere with 1 to 2 mg of sample. Thermal decomposition kinetic studies were also undertaken on a Mettler Toledo SDT Q600 V20.9 Build 20, operating at a heating rate ranging from 4 °C min−1 to 10 °C min−1. The Impact sensitivity of both compounds was determined by BAM fall hammer method using 2 kg drop weight. The results are reported in terms of height for 50% probability of explosion (h50%) of the sample. The friction sensitivity of the compounds was measured on a Julius Peter's apparatus till there was no explosion/ignition in five consecutive test samples at that weight. The results obtained for impact and friction sensitivity were within the uncertainty limits of ±5 cm and ±0.2 kg, respectively. The energetic binders, Glycidyl azide polymer (GAP) (Mn− ≈ 2000), and 3,3-bis-azidomethyl oxetane (BAMO) (Mn− ≈ 3500), used for compatibility study were taken from High Energy Materials Research Laboratory (HEMRL), Pune, India.
Caution! As organic azides are classified as toxic and highly explosives, proper safety measures must be taken at all times. Although in our case no difficulties in handling these energetic materials, small-scale reaction is always preferred.
Typical procedure for the preparation of azido ester
(a) Method A. To a solution of halo alcohol in DMF, NaN3 was added to that with cooling. The reaction mixture was slowly warmed to 85 °C on oil bath with stirring for18 h. The reaction mixture was then poured into ice water and extracted with ethyl acetate (3 × 30 mL). The ethyl acetate extract was washed with brine (3 × 30 mL) and then dried over Na2SO4. Evaporation of solvents furnished crude which was purified by silica-gel column chromatography using 20% ethyl acetate–hexane to deliver pure product as pale yellow liquid 2.
(b) Method B. To a solution of 1,3-dichloro propan-2-ol (1.2134 g, 9.41 mmol) in DMF (15 mL), NaN3 (0.7337 g, 11.29 mmol) was added to that with cooling. The reaction mixture was slowly warmed to 40 °C on oil bath with stirring for 24 h. The reaction mixture was then poured into ice water and extracted with ethyl acetate (3 × 30 mL). The ethyl acetate extract was washed with brine (3 × 30 mL) and then dried over Na2SO4. Evaporation of solvents furnished crude which was purified by silica-gel column chromatography using 20% ethyl acetate–hexane to deliver pure product as pale yellow liquid 2 (1.33 g, 75%).
(c) Method C. To a solution of 1,3-dichloro propan-2-ol (0.8213 g, 6.37 mmol) in DMSO/H2O (80
:20%) (8 mL DMSO: 2 mL H2O), NaN3 (0.4966 g, 7.64 mmol) was added to that with cooling. The reaction mixture was slowly warmed to 40 °C on oil bath with stirring for 24 h. The reaction mixture was then poured into ice water and extracted with ethyl acetate (3 × 30 mL). The ethyl acetate extract was washed with brine (3 × 30 mL) and then dried over Na2SO4. Evaporation of solvents furnished crude which was purified by silica-gel column chromatography using 20% ethyl acetate–hexane to deliver pure product as pale yellow liquid 2 (0.65 g, 72%).
(a) Method A. To a solution of carboxylic acid in CH2Cl2, N,N′- dicyclohexylcarbodiimide, 1,3-di azido propan-2-ol and 4-(dimethylamino)pyridine were added. The reaction mixture was stirred overnight at room temperature, filtered, washed with 1 M HCl (3 × 10 mL), 20% aqueous NaHCO3 (3 × 10 mL) and water (3 × 10 mL). The organic layer was dried (MgSO4), filtered and evaporated in vacuo. Evaporation of solvents furnished crude which was purified by silica-gel column chromatography using ethyl acetate–hexane to deliver pure product. By following this method compounds 6a–m has been synthesized.
(b) Method B. To a solution of 3-trifluoro methyl-4-nitro benzoic acid (0.2214 g, 0.94 mmol) in CH2Cl2 (30 mL), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC hydrochloride) (0.1805 g, 0.94 mmol) and 1,3-di azido propan-2-ol (0.1606 g, 1.13 mmol) were added. The reaction mixture was stirred overnight at room temperature, washed with 20% aqueous NaHCO3 (3 × 10 mL), water (3 × 10 mL) and brine solution (3 × 10 mL). The organic layer was dried (MgSO4), filtered and evaporated in vacuo. Evaporation of solvents furnished final product as pale yellow liquid (6b) (0.25 g, 75%).
1,3-dichloro propan-2-ol (1.933 g, 14.99 mmol), DMF (15 mL), and NaN3 (2.922 g, 44.95 mmol) were used. Purification by 20% ethyl acetate–hexane to deliver pure product (2) as pale yellow liquid (1.81 g, 85%). 1H NMR (CDCl3, 300 MHz) δ 3.26 (1H, d, J = 4.8 Hz), 3.41 (4H, d, J = 6.2 Hz), 3.93 (1H, quin, J1 = 10.5 Hz J2 = 5.2 Hz).13C NMR (CDCl3, 75 MHz) δ 53.6, 69.4 ppm.
Conclusions
A simple straight forward approach has been adopted for synthesis of several novel di azido, tetra azido and hexa azido ester plasticizers from readily available commercial reagents. All these compounds exhibit good high heat release capabilities, high nitrogen content and insensitivity towards impact and friction. All azido-esters exhibit positive heats of formation. It has also been noticed that substitution of azido-ester (–CO–O–CH(CH2N3)2) group increases the heats of formation of compounds by about 200–250 kJ mol−1. Thus, substitution of azido-esters group in compounds can be a valuable strategy for improving heats of formation of compounds. All synthesized azido esters except solids, show promising results with respect to compatibility with energetic binders. All these properties make these azido esters as potential candidate for their application as energetic plasticizers.A simple straight forward approach has been adopted for synthesis of several novel di azido, tetra azido and hexa azido ester plasticizers from readily available commercial reagents. All these compounds exhibit good high heat release capabilities, high nitrogen content and insensitivity towards impact and friction. All azido-esters exhibit positive heats of formation. It has also been noticed that substitution of azido-ester (–CO–O–CH(CH2N3)2) group increases the heats of formation of compounds by about 200–250 kJ mol−1. Thus, substitution of azido-esters group in compounds can be a valuable strategy for improving heats of formation of compounds. All synthesized azido esters except solids, show promising results with respect to compatibility with energetic binders. All these properties make these azido esters as potential candidate for their application as energetic plasticizers.
Acknowledgements
The authors would like to thank the Vice Chancellor, DIAT for providing the infrastructural facilities. Financial support from ER&IPR, DRDO, New Delhi through “DRDO-DIAT programme on Nano Materials” is gratefully acknowledged. DK thanks CSIR, New Delhi for providing fellowship. We thank Dr A. K. Sikder and Ms. Suman Pawar of EMR Division, HEMRL, Pune for providing support towards thermal characterization.References
- S. Bräse, C. Gil, K. Knepper and V. Zimmermann, Angew. Chem., Int. Ed., 2005, 44, 5188 CrossRef PubMed.
- T. M. Klapötke, Chemistry of High Energy Materials, Walter de Gruyter, Berlin/New York, 2012 Search PubMed.
- S. Bräse and K. Banert, Organic Azides Syntheses and Applications, John Wiley & Sons, UK, 2010 Search PubMed.
- K. Banert, Y. H. Joo, T. Rüffer, B. Walfort and H. Lang, Angew. Chem., Int. Ed., 2007, 46, 1168 CrossRef CAS PubMed.
- D. M. Badgujar, M. B. Talawar, S. N. Asthana and P. P. Mahulikar, J. Hazard. Mater., 2008, 151, 289 CrossRef CAS PubMed.
- M. B. Talawar, R. Sivabalan, T. Mukundan, H. Muthurajan, A. K. Sikder, B. R. Gandhe and A. S. Rao, J. Hazard. Mater., 2009, 161, 589 CrossRef CAS PubMed.
- J. P. Agrawal and R. D. Hodgson, Organic Chemistry of Explosives, John Wiley & Sons, UK, 2007 Search PubMed.
- M. B. Frankel and E. R. Wilson, J. Chem. Eng. Data, 1981, 26(2), 219 CrossRef CAS.
- E. F. Witucki and M. B. Frankel, J. Chem. Eng. Data, 1979, 24, 247 CrossRef CAS.
- E. F. Witucki, E. R. Wilson, J. E. Flanagan and M. B. Frankel, J. Chem. Eng. Data, 1982, 28(2), 285 CrossRef.
- E. F. Witucki and M. B. Frankel, J. Chem. Eng. Data, 1982, 27, 94 CrossRef CAS.
- F. D. Marsh, J. Org. Chem., 1972, 37, 2966 CrossRef CAS.
- G. Ampleman, Synthesis of diazido terminated energetic plasticizer, U.S. Patent 5,124,463, June 23, 1992.
- P. R. Dave, R. G. Duddu, R. Damavarapu, N. Gelber, K. Yang and C. R. Surapaneni, Polyazido compounds, U.S. Patent US 7,109,359 B1, September 19, 2006.
- K. Ghosh, J. Athar, S. Pawar, B. G. Polke and A. K. Sikder, J. Energ. Mater., 2012, 30, 107 CrossRef CAS.
- H. Gao and J. M. Shreeve, Chem. Rev., 2011, 111, 7377 CrossRef CAS PubMed.
- S. Garg and J. M. Shreeve, J. Mater. Chem., 2011, 21, 4787 RSC.
- C. Ye, G. L. Gard, R. W. Winter, R. G. Syvret, B. Twamley and J. M. Shreeve, Org. Lett., 2007, 9, 3841–3844 CrossRef CAS PubMed.
- R. P. Singh, R. D. Verma, D. T. Meshri and J. M. Shreeve, Angew. Chem., Int. Ed., 2006, 45, 3584 CrossRef CAS PubMed.
- G. H. Tao, B. Twamley and J. M. Shreeve, J. Mater. Chem., 2009, 19, 5850 RSC.
- T. M. Klapötke and J. Stierstorfer, J. Am. Chem. Soc., 2009, 131, 1122 CrossRef PubMed.
- R. Wang, H. Xu, Y. Guo, R. Sa and J. M. Shreeve, J. Am. Chem. Soc., 2010, 132, 11904 CrossRef CAS PubMed.
- G. Wypych, Handbook of Plasticizers, Chem Tec Publishing, Toronto, Canada, 2004 Search PubMed.
- D. Kumari, R. Balakshe, S. Banerjee and H. Singh, Res. Rev.: J. Chem., 2012, 2, 240 CrossRef.
- H. G. Ang and S. Pisharath, Energetic Polymers: Binders and Plasticizers for Enhancing Performance, Wiley-VCH, 2012 Search PubMed.
- D. Kumari, K. D. B. Yamajala, H. Singh, R. R. Sanghavi, S. N. Asthana, K. Raju and S. Banerjee, Propellants, Explos., Pyrotech., 2013, 38, 805 CrossRef CAS PubMed.
- K. Ghosh, C. S. Pant, R. Sanghavi, S. Adhav and A. Singh, J. Energ. Mater., 2009, 27, 40–50 CrossRef CAS.
- F. Schedlbauer, Propellants, Explos., Pyrotech., 1992, 17, 164–170 CrossRef CAS PubMed.
- R. R. Sanghavi, P. J. Kamale, M. A. R. Shaikh, S. D. Shelar, K. S. Kumar and A. Singh, J. Hazard. Mater., 2007, 143, 532 CrossRef CAS PubMed.
- R. R. Sanghavi, V. H. Khire, T. K. Chakraborthy and A. Singh, Propellants, Explos., Pyrotech., 2006, 31, 318 CrossRef CAS PubMed.
- E. R. Wilson and M. B. Frankel, J. Chem. Eng. Data, 1982, 27, 472 CrossRef CAS.
- E. R. Wilson and M. B. Frankel, J. Org. Chem., 1985, 50, 3211 CrossRef CAS.
- C. S. Pant, R. M. Wagh, J. K. Nair, G. M. Gore, M. Thekkekara and S. Venugopalan, Propellants, Explos., Pyrotech., 2007, 32, 461–467 CrossRef CAS PubMed.
- C. S. Pant, R. M. Wagh, J. K. Naira and T. Mukundana, J. Energ. Mater., 2006, 24, 333 CrossRef CAS.
- C. S. Pant, R. M. Wagh, J. K. Nair, G. M. Gore and S. Venugopalan, Propellants, Explos., Pyrotech., 2006, 31, 477 CrossRef CAS PubMed.
- Q. Shaojun, F. Huiqing, G. Chao, Z. Xiaodong and G. Xiaoxian, Propellants, Explos., Pyrotech., 2006, 31, 205 CrossRef PubMed.
- D. Drees, D. Loffel, A. Messmer and K. Schmid, Propellants, Explos., Pyrotech., 1999, 24, 159 CrossRef CAS.
- D. Kumari, H. Singh, M. Patil, W. Thiel, C. S. Pant and S. Banerjee, Thermochim. Acta, 2013, 562, 96 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS.
- (a) L. Liang, H. Haung, K. Wang, C. Bian, J. Song, L. Ling, F. Zhaoc and Z. Zhou, J. Mater. Chem., 2012, 22, 21954 RSC; (b) G. Wang, X. Gong, H. Du, Y. Liu and H. Xiao, J. Phys. Chem. A, 2011, 115, 795 CrossRef CAS PubMed; (c) L. Türker and T. Atlar, J. Hazard. Mater., 2009, 162, 193 CrossRef PubMed; (d) D. E. Chavez, M. A. Hiskey, D. L. Naud and D. Parrish, Angew. Chem., Int. Ed., 2008, 47, 8307 CrossRef CAS PubMed.
- M. H. Huynh, M. A. Hiskey, D. E. Chavez, D. L. Naud and R. D. Gilardi, J. Am. Chem. Soc., 2005, 127, 12537 CrossRef CAS PubMed.
- M. E. Brown, Thermochim. Acta, 1989, 148, 521 CrossRef.
- S. Pisharath and H. G. Ang, Thermochim. Acta, 2007, 459, 26 CrossRef CAS PubMed.
- S. M. Pourmortazavi, M. R. Nasrabadi, I. Kohsari and S. S. Hajimirsadeghi, J. Therm. Anal. Calorim., 2012, 110, 857 CrossRef CAS.
- B. Roduit, L. Xia, P. Folly, B. Berger, J. Mathieu, A. Sarbach, H. Andres, M. Ramin, B. Vogelsanger, D. Spitzer, H. Moulard and D. Dilhan, J. Therm. Anal. Calorim., 2008, 93, 143 CrossRef CAS.
- J. A. F. F. Rocco, J. E. S. Lima, A. G. Frutuoso, K. Iha, M. Ionashiro, J. R. Matos and M. E. V. Suárez-Iha, J. Therm. Anal. Calorim., 2004, 75, 551 CrossRef CAS.
- H. E. Kissinger, Anal. Chem., 1957, 29, 1702 CrossRef CAS.
- P. Budrugeac and E. Segal, J. Therm. Anal. Calorim., 2007, 88, 703 CrossRef CAS.
- R. L. Blaine and H. E. Kissinger, Thermochim. Acta, 2012, 540, 1 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR (1H, 13C, 19F, COSY, DEPT and HMBC) spectra S2–S18 (Fig. S1 to S34), DSC & TGA Curves S19–S30 (Fig. S35 to S92), Computational Studies data S31–S44. See DOI: 10.1039/c4ra06530a |
| This journal is © The Royal Society of Chemistry 2014 |