
Switching the thermodynamics of MgH2 nanoparticles through polystyrene stabilisation and oxidation†
Eki J. Setijadiab,
Cyrille Boyerb and
Kondo-Francois Aguey-Zinsou*a
aMERLin Group, School of Chemical Engineering, The University of New South Wales, Sydney, NSW 2052, Australia. E-mail: f.aguey@unsw.edu.au; Fax: +61 (0)2 938 55966; Tel: +61 (0)2 938 57970
bCentre for Advanced Macromolecular Design, School of Chemical Engineering, The University of New South Wales, Sydney, NSW 2052, Australia
First published on 19th August 2014
Abstract
Magnesium is a promising material for hydrogen storage purposes; however, modifying the thermodynamic properties of the magnesium/hydrogen reaction remains important for practical application. Herein, we report an exciting finding that allows switching the thermodynamic properties of polystyrene stabilised magnesium nanoparticles via simple exposure to air. The magnesium nanoparticles stabilised with polystyrene were synthesised by direct hydrogenolysis of di-n-butylmagnesium. Polystyrene was found to significantly influence the nucleation and growth process leading to nanoparticles of ∼100 nm with thermodynamic properties similar to that of bulk magnesium. However, upon partial oxidation and hydrogen cycling, these nanoparticles were found to undergo a significant morphological reconstruction and particle size reduction leading to a drastic shift in thermodynamic properties with both enthalpy and entropy decreasing to 52.3 ± 3.2 kJ mol−1 H2 and 101.3 ± 4.5 J mol−1 K−1 H2, respectively. Such a shift in thermodynamics demonstrates the possibility of tuning the thermodynamics of magnesium through the use of appropriate external factors such as partial oxidation while maintaining a reasonable storage capacity.
1. Introduction
Magnesium hydride (MgH2) still remains as an ideal candidate for hydrogen storage purposes owing its high hydrogen storage capacity (7.6 wt%), abundance and non-toxic nature. However, the slow hydrogen sorption kinetics and the need for high temperatures to effectively store hydrogen remains the limits to its practical use.1,2 Many efforts have been made to tackle these problems and improvement through mechanical milling has been the dominant approach for a few decades.3,4 Indeed, nanostructuring approaches have led to substantial improvements especially of the kinetics due to an increase in surface area and catalysts at grain boundaries. Some theoretical work also showed the possibility of thermodynamic improvements through a nanosizing approach, however with very small magnesium particles <3 nm.5–7 Nonetheless, we have recently shown that achieving particles as small as 3 nm was not necessary and significant shifts in thermodynamics could be observed for much larger nanoparticles ∼25 nm.8Another issue associated with the use of MgH2 nanoparticles is their greater sensitivity to oxidation as compared to bulk magnesium. In general, a short exposure to air, moisture or oxidising agents will be detrimental to the storage capacity with the formation of a passivating oxide layer (3–4 nm thick) leading to capacity loss and poorer kinetics.9 However, some reports have also claimed kinetics improvements upon a very small oxidation of magnesium surfaces,10,11 and we have shown the benefit of MgO in improving the hydrogen sorption properties of MgH2 through ball-milling.12 Accordingly, effective stabilisation methods are important in a nanosizing approach not only to prevent nanoparticles self-agglomeration and sintering during hydrogen cycling but also to protect magnesium nanoparticles against excessive oxidation. Agglomeration can be reduced by nanoconfinement strategies and several studies have reported the use of porous structures including carbon to host magnesium nanoparticles.13–16 However, the inherent difficulty of entirely filling the porosity of the host structure often leads to a drastic reduction in storage capacity (<2 wt%). Moreover, nanoconfined magnesium particles often display very slow kinetics possibly due to the long diffusion ranges involved within porous matrixes.
Surfactants and other organic ligands offer an alternative approach to stabilise nanoparticles and prevent oxidation. However, organic stabilisers should be carefully selected to avoid any side reactions with magnesium and their thermal decomposition during hydrogen cycling.17 Hence, although magnesium nanoparticles stabilised by tetrabutylammonium (TBAB) showed hydrogen release below 100 °C, only a few cycles could be achieved owing to the thermal degradation of TBAB.18 Similarly, 5 nm MgH2 nanoparticles stabilised with poly(methyl methacrylate) (PMMA) achieved a few hydrogen cycles at 200 °C with around 4 wt% capacity.19 The degradation of the cycling properties of PMMA stabilised magnesium nanoparticles may be due to the reactivity of the methacrylate function with magnesium and the relatively low thermal stability of PMMA degrading from 250 °C. However, the stabilisation with PMMA appeared to be an effective strategy to prevent oxidation of the 5 nm magnesium nanoparticles even when exposed to air although the cycling and thermodynamic properties of the oxidised material were not reported.19,20
Based on these preliminary results and the potential role of oxidation in improving the hydrogen kinetics of magnesium, we have investigated the use of polystyrene to stabilise against oxidation magnesium nanoparticles generated by the hydrogenolysis of di-n-butylmagnesium. The later was found to lead to magnesium nanoparticles with particle sizes ranging from 25 to 50 nm.21,22 Polystyrene with a carbon chain free from potentially reactive functional groups was expected to lead to the formation of smaller and stabilised magnesium nanoparticles upon hydrogen cycling and provide further protection against oxidation and thermal degradation of the magnesium nanoparticles upon hydrogen cycling. Indeed, polystyrene like PMMA has low oxygen permeability (2.4 and 3.3 Barrer, respectively)23 and thus should lead to effective protection against oxidation while enabling hydrogen sorption. In addition, polystyrene only starts to decompose at 350 °C,24 and thus provides a better temperature window for cycling magnesium as compared to PMMA which decomposes from 200 °C. We indeed found that magnesium nanoparticles with enhanced storage capacity were obtained upon the use of polystyrene as a stabiliser. Furthermore, polystyrene stabilised magnesium nanoparticles did not show any degradation of storage capacity upon cycling which allowed the determination of the evolution of the thermodynamics and kinetics properties of this hybrid material upon exposure to air and oxidation.
2. Experimental section
All material handling, weighing, loading and washing was performed in a glove box filled with high purity argon (O2 and H2O <1 ppm) from LC Technology.2.1. Materials and synthesis
Di-n-butylmagnesium (1.0 M in heptane and up to 1 wt% triethylaluminum as viscosity reducer) and anhydrous cyclohexane were obtained from Sigma-Aldrich. Polystyrene (PSTN) with a low molecular weight (MW = 5000) to ensure solubility in cyclohexane was synthesized following previous procedures.25Di-n-butylmagnesium was dried under vacuum for solvent removal. The powder obtained (2.00 g) was resuspended in cyclohexane (100 mL) with and PSTN (0.400 g). The hydrogenolysis of di-n-butylmagnesium was then carried out at 180 °C for 24 h under a hydrogen pressure of 3 MPa. The resulting grey precipitates (MgH2/PSTN) was collected by centrifugation, washed several times with fresh cyclohexane and dried under vacuum at room temperature (0.577 g, 96% yield). This method was repeated without PSTN to lead MgH2 only as a reference material (0.314 g, 85% yield). The materials were oxidised by exposing the powder obtained to air for 24 h. All further characterisations were then performed under a controlled atmosphere.
2.2. Methods
Transmission Electron Microscopy (TEM) and Energy Dispersive X-ray spectroscopy (EDX) analysis were performed on a Phillips CM200 microscope operating at 200 keV. The materials were dispersed in THF and deposited onto a carbon coated copper grid. Materials preparation was carried out in a glove box under high purity Ar. The materials were exposed to air during transfer to the microscope.Structural characterization was performed using a Phillips X'pert X-ray Diffraction system (XRD) operating at 40 mA and 4 kV using monochromated Cu-Kα radiation (λ = 1.541 Å) – step size = 0.01, 0.02 or 0.05, time per step = 10 or 20 s per step. The materials were protected against oxidation from air by a Kapton foil.
The chemical surface composition of the nanoparticles synthesized was determined by X-ray Photoelectron Spectroscopy (XPS) using an EscaLab 220-IXL from VG Scientific. The instrument was operated with a monochromated Al-Kα radiation at 1486.60 eV and a power source of 120 W. A spot size of 0.5 mm in diameter with a pass energy of 100 eV was used for wide scans and a pass energy of 20 eV was used for narrow scans of particular elemental peaks. The materials were exposed to air during transfer to the instrument.
Investigations on thermal stability and hydrogen desorption profiles were performed by Thermal Gravimetric Analysis (TGA) and Differential Scanning Calorimetry (DSC) coupled with Mass Spectrometry (MS) using a Mettler Toledo TGA/DSC 1 and an OmniStar Mass Spectrometer. The instruments were installed in an argon filled glove box (<1 ppm O2 and H2O) to avoid any oxidation of the materials. Typically, 4 mg of material was used and the characterization was run with a heating rate of 10 °C min−1 under a flow of 40 mL min−1 of high purity argon (99.9999%).
Hydrogen absorption and desorption kinetics, as well as the Pressure-Composition Isotherms (PCI) were measured on an automatic Sieverts apparatus (Advanced Materials Corporation). For the kinetic measurements, a hydrogen pressure of 3 MPa was used for absorption and a pressure of 0.05 MPa for desorption. Prior PCI measurements the materials were put under vacuum (0.01 MPa) for 12 h at 300 °C and then cycled 3 times at the same temperature. The PCI measurements were performed with a step size of 0.04 MPa at various different temperatures of 280, 300, 310, and 325 °C to determine the equilibrium plateau pressure (Peq). From these PCI measurements values of enthalpy and entropy were obtained from a Van't Hoff plot following the equation ln(Peq) = (ΔH/R)(1/T) − ΔS/R. The latter was determined by plotting the logarithm of Peq at a given temperature versus the inverse of the measurement temperature. Only the PCI for absorption are reported here.
3. Results and discussion
The hydrogenolysis of di-n-butylmagnesium proceeds according to the simplified path (1) and under mild temperature conditions since the decomposition of di-n-butylmagnesium occurs from 110 °C with excessive degradation above 150 °C.| (C4H9)2Mg → 2C4H8(g) + MgH2(s) | (1) |
This decomposition leads to the direct formation of MgH2 even under an inert atmosphere.21,26 However, for achieving fast desorption kinetics and high storage capacity, the hydrogenolysis should be performed under hydrogen pressure or in a non-polar solvent inert toward magnesium such as cyclohexane.22 Accordingly, to facilitate the stabilization of magnesium by PSTN, the synthesis was performed in cyclohexane.
3.1. Properties of the magnesium nanoparticles synthesised
The morphology of the new composite, MgH2/PSTN, was investigated by TEM and compared to that of the MgH2 resulting from the hydrogenolysis process of di-n-butylmagnesium only (material noted MgH2/C). As previously reported, the decomposition of di-n-butylmagnesium in cyclohexane resulted in the formation of nanoparticles with a size ranging from 25 to 50 nm (Fig. 1a). Surprisingly, the use of PSTN led to the formation of bigger particles (∼100 nm) with a rectangular shape (Fig. 1b and Table 1). These nanoparticles correspond to magnesium as confirmed by EDS analysis (Fig. 1C insert). Furthermore, by XRD analysis the crystalline nature of both materials was established as that of β-MgH2 (Fig. 2). An aluminium phase was also detected by XRD, and this originated from the small amount of triethylaluminum in the di-n-butylmagnesium solution. Using the Scherrer formula the crystallite size was determined for both materials and found to be relativity similar ∼15 nm although larger particles were synthesised with PSTN (Table 1). | ||
| Fig. 1 TEM images of: (a) MgH2/C and (b) MgH2/PSTN as-synthesized and after hydrogen absorption/desorption cycling at 300 °C (c) MgH2/C including EDX analysis insert and (d) MgH2/PSTN. | ||
| Materials | Particle morphology/size (nm) | Crystallite size (nm ± 3) | |
|---|---|---|---|
| After synthesis | After cycled | ||
| MgH2/C | Round like, 25–50 | 12 | 28 |
| MgH2/C oxidised | Round like, 25–50 | 12 | 30 |
| MgH2/PSTN | Rectangular, ∼50 × 100 | 18 | 32 |
| MgH2/PSTN oxidised | Rectangular, ∼50 × 100 | 16 | 36 |
 | ||
| Fig. 2 XRD patterns of MgH2/C and MgH2/PSTN as-synthesized and after hydrogen desorption. The diffraction patterns after hydrogen absorption are identical to that of the as-synthesised materials. | ||
Hydrogen desorption followed by TGA/MS from the as-synthesised MgH2/C was found to start from 300 °C with a main peak at 343 °C (Fig. 3a). In comparison, desorption from MgH2/PSTN started at a slightly higher temperature of 330 °C with a main peak at 356 °C. This shift in temperature for MgH2/PSTN may be due to the larger magnesium particles formed and/or the polystyrene coating. Indeed by MS, fragments corresponding to the decomposition of polystyrene were detected from 380 °C proving that some polymer remained within the material (Fig. 3b). The remaining polymer may be bonded to the magnesium nanoparticles since its decomposition temperature is higher than that of pristine polystyrene, i.e. from 320 °C instead of the 380 °C observed for MgH2/PSTN (Fig. 3b and S1†). Magnesium is known to react with a range of organic molecules to form organomagnesium compounds;27 hence the formation of magnesium–polystyrene bonds may be possible. Further determination of the storage capacity of the materials and hydrogen sorption kinetics was performed by PCI measurements. Herein, only desorption kinetics are reported. Hydrogen absorption is an exothermic process usually fast while the endothermic desorption of hydrogen in magnesium remains the main problem. At 300 °C, MgH2/C was found to release 5 wt% of hydrogen in 70 min (Fig. 4), which is significantly faster than the kinetics of ball-milled magnesium (>200 min) measured under the same conditions.8 In comparison, the desorption kinetics of MgH2/PSTN were found to be slower with full hydrogen release achieved in 150 min. This slower desorption kinetics of MgH2/PSTN is in agreement with the hydrogen desorption profiles measured by TGA/MS (Fig. 3), and may be due to the larger particles size of MgH2/PSTN compared to MgH2/C (Table 1) and/or different activation energy (Ea). It is well acknowledged that smaller particles would lead to faster kinetics due to shorter of diffusion distances, hence slower kinetics could be expected for MgH2/PSTN. However, the determination of Ea by the Kissinger method for both materials revealed higher activation energy for MgH2/PSTN as compared to MgH2/C (Table 2). Hence, the slower kinetics observed for MgH2/PSTN may predominantly be due to slower recombination rates of hydrogen at the surface of the magnesium particles covered with PSTN. Furthermore, XRD analysis confirmed the conversion of the tetragonal β-MgH2 into the hexagonal Mg phase upon hydrogen release proving that hydrogen was effectively stored upon hydrogen cycling (Fig. 2). Remarkably these materials did not show any degradation of storage capacity upon repeated cycles and successive PCI measurements proving the better stability of PSTN as compared to PMMA. Analysis by TEM of the materials after hydrogen cycling also revealed no significant changes in the morphology of MgH2/C and MgH2/PSTN (Fig. 1c and d).
 | ||
| Fig. 3 Hydrogen desorption profiles as measured by TGA/MS of (a) MgH2/C and (b) MgH2/PSTN as-synthesised, after hydrogen cycling, and after oxidation for 24 h in air. | ||
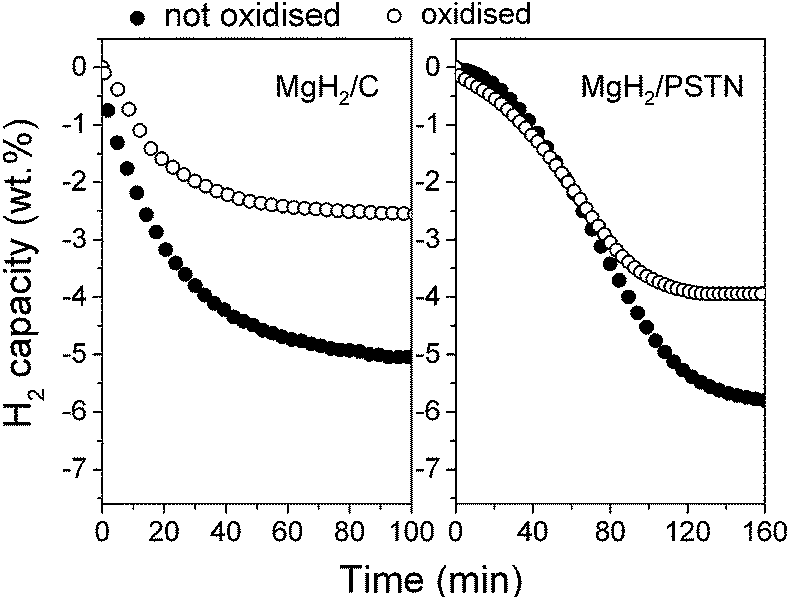 | ||
| Fig. 4 Hydrogen desorption kinetics measured at 300 °C for (a) MgH2/C and (b) MgH2/PSTN, as synthesised and after oxidation for 24 h in air. The kinetics were found to be stable even after PCI measurements (ESI Fig. S5†). | ||
| Materials | ΔH (kJ mol−1 H2) | ΔS (J mol−1 K−1 H2) | Ea (kJ mol−1 H2) |
|---|---|---|---|
| MgH2/C | −51.5 ± 3.1 | 99.5 ± 4.5 | 133.0 ± 5.0 |
| MgH2/C exposed | −52.4 ± 3.6 | 101.8 ± 3.1 | 189.4 ± 4.9 |
| MgH2/PSTN | −68.1 ± 3.2 | 127.2 ± 4.1 | 171.3 ± 3.9 |
| MgH2/PSTN exposed | −52.3 ± 3.2 | 101.3 ± 4.5 | 196.2 ± 4.1 |
| Ball-milled MgH2 (ref. 8) | −75.2 ± 1.8 | 139.0 ± 3.0 | — |
Surprisingly, the storage capacity of MgH2/PSTN was found to be higher than that of MgH2/C, i.e. 5.8 instead of 5 wt%, respectively (Fig. 4). Further investigations by TGA/MS showed that at 300 °C, only hydrogen was released from both materials with additional decomposition of the polymer for MgH2/PSTN still occurring from 380 °C after hydrogen release (Fig. 3). Hence, the extra storage capacity of MgH2/PSTN was attributed to hydrogen release only. Such behaviour, observed for the first time with magnesium may be due to particle size effects. Indeed, similar effects have been observed with Pd for decreasing particle sizes. Hence, the narrowing of the equilibrium plateau pressure – or miscibility gap corresponding to the coexistence of the α and β phase – observed with decreasing Pd particle sizes, led to a reduction of the storage capacity, e.g. from 0.6–0.7 to 0.3 H/Pd for bulk and 3 nm Pd nanoparticles, respectively.28 This has been explained by a reduction of the nanoparticle volume that will transform into the hydride phase (β phase) and the associated decrease of both enthalpy and entropy leading to weaker Pd–H bond strengths and also greater freedom of the hydrogen atoms within Pd nanoparticles as compared to bulk Pd. Further determination of the thermodynamic of MgH2/C and MgH2/PSTN by PCI measurements and associated Van't Hoff plot (ESI Fig. S2, S3† and 5) revealed a significant decrease of both the enthalpy (ΔH) and entropy (ΔS) of MgH2/C as compared to MgH2/PSTN and bulk MgH2 (Table 2). In comparison to bulk magnesium, this decrease is of ΔH = 23.7 kJ mol−1 H2 and ΔS = 39.5 kJ mol−1 K−1 H2 for MgH2/C, and ΔH = 7.1 kJ mol−1 H2 and ΔS = 8.8 kJ mol−1 K−1 H2 for MgH2/PSTN. Hence, while the thermodynamic properties of MgH2/PSTN are relatively closed to that of bulk magnesium, the significant decrease in ΔH for MgH2/C compensated by a decrease in ΔS would corroborate the resulting decrease in storage capacity as observed with Pd nanoparticles. It is noteworthy although ΔH significantly decreased for MgH2/C the likewise reduction in ΔS also led to an overall reduction of the desorption temperature Tdes = ΔH/ΔS of 24 °C only at 1 bar hydrogen pressure compared to bulk magnesium.
 | ||
| Fig. 5 Van't Hoff plots of MgH2/C and MgH2/PSTN as synthesized and oxidised. Equilibrium pressures were obtained from the PCI measurements. | ||
3.2. Effect of oxidation from air exposure
Magnesium nanoparticles are expected to be highly flammable once exposed to air. However, the materials were found to be stable with no apparent degradation after 24 h. Indeed, TEM analysis did not show any significant evolution of particles morphology (Fig. 6). XRD characterisation did not either show any formation of MgO phases with diffraction patterns identical to the as-synthesised materials (Fig. 2). However, further characterisation by XPS analysis revealed that all materials were oxidised to some extent (Fig. 7). Indeed, the O 1s peak showed two different chemical states at 531.7 and 529.6 eV attributed to magnesium hydroxide and oxide, respectively.29,30 Nonetheless, the oxide layer was thin enough to allow the detection of metallic magnesium underneath as shown by the strong Mg 2p peak at 49.9 eV alongside a peak at 51.9 eV attributed to magnesium hydroxide and/or oxide (Fig. 8a).29,30 It is noteworthy that even after 24 h of exposure to air the oxide layer did not seems to significantly grow since metallic magnesium was still detected for both oxidised materials. This is in agreement with previous reports claiming the rapid formation of passivating oxide layer of 3–4 nm preventing further oxidation.9 | ||
| Fig. 6 TEM images of: (a) MgH2/C and (b) MgH2/PSTN as-synthesized and oxidised for 24 h in air and associated images after hydrogen cycling at 300 °C (c) MgH2/C and (d) MgH2/PSTN. | ||
 | ||
| Fig. 7 XPS peak profiles (a) Mg 2p and (b) O 1s of MgH2/C and MgH2/PSTN as-synthesized and oxidised. | ||
 | ||
| Fig. 8 XRD patterns of MgH2/C and MgH2/PSTN as-synthesized and oxidised after hydrogen cycling, PCI measurements and absorption. | ||
From TGA/MS analysis (Fig. 3), the hydrogen desorption profile of MgH2/C shifted to slightly higher temperatures with a main peak at ∼350 °C instead of 343 °C (Fig. 3a). However, a bigger shift of 24 °C was observed for MgH2/PSTN and this may due to the oxide layer retarding hydrogen release. The determination of the storage capacity by PCT measurements also confirmed the oxidation of both materials. MgH2/C lost half its storage capacity, while MgH2/PSTN retains almost 70% of its original storage capacity (Fig. 4). Surprisingly, desorption kinetics were also found to be somewhat faster as compared to the non-oxidised materials with 95% of hydrogen release from MgH2/C achieved in 50 min for the oxidised material instead of 70 min, for example (Fig. 4). Various reports have suggested partial oxidation of magnesium surfaces could effectively catalyse hydrogen sorption.10,11 However, the determination of the activation energy (Ea) by the Kissinger's method showed a significant increase in Ea for the materials exposed to air (Table 2). For example, Ea was found to increase from 133.0 ± 5.0 kJ mol−1 H2 to 189.4 ± 4.9 kJ mol−1 H2 for MgH2/C as synthesised and exposed to air, respectively. Assuming that the oxidation of magnesium particles will lead to the growth of a uniform surface oxide layer resulting in a smaller magnesium core, it is more likely that the faster kinetics are due to the smaller size of the active magnesium core within particles.
The thermodynamic properties of the oxidised materials were also determined from the PCI measurements and the associated Van't Hoff plots are reported in Fig. 5. For MgH2/C no significant change in enthalpy and entropy was observed. However, both ΔH and ΔS for MgH2/PSTN oxidised were found to drastically decrease to 52.3 ± 3.2 kJ mol−1 H2 and 101.3 ± 4.5 kJ mol−1 K−1 H2, respectively. This is equivalent to the values observed for MgH2/C and the evolution of changes as function of particle sizes previous reported.8 Hence, based on this result only a significant decrease in particle size for MgH2/PSTN oxidised should have occurred during cycling. Indeed, TEM analysis revealed a change in morphology and much smaller magnesium particles sizes ∼25–50 nm for MgH2/PSTN oxidised and cycled (Fig. 6d). Further characterisation by XRD of the oxidised materials after hydrogen cycling finally showed the formation of a MgO phase for MgH2/C only (Fig. 8). For MgH2/PSTN the oxide layer may remain amorphous or as a finely dispersed phase, since the oxidation of MgH2/PSTN resulted in a significant reconstruction of the magnesium particles upon hydrogen cycling (Fig. 6d). The driving forces for such a reconstruction remain unclear and are most likely driven by PSTN/magnesium oxide interface at the surface of the magnesium nanoparticles and possible associated stress upon hydrogen absorption/desorption. Decrepitation of metal hydrides is a well know phenomenon for microsized powders,31,32 but to the best of our knowledge this is the first time that it is observed for nanosized magnesium particles. For microsized powered this leads to increase surface areas and thus enhance hydrogen kinetics, at the nanosize it obviously provides an additional path to modify the thermodynamics properties of the magnesium/hydrogen reaction.
4. Conclusions
The thermal decomposition di-n-butylmagnesium with polystyrene as a stabiliser was found to lead to the formation of relatively large magnesium nanoparticles (∼100 nm) with a rectangular morphology. These nanoparticles had a higher storage capacity as compared to the nanoparticles (25–50 nm) generated from the hydrogenolysis of di-n-butylmagnesium alone but their thermodynamic properties was close to that of bulk magnesium, i.e. ΔH = −68.1 ± 3.2 kJ mol−1 H2 and ΔS = 127.2 ± 4.1 J mol−1 K−1 H2. The difference in storage capacity was attributed to particle size differences and the ability of extra storage capacity within larger nanoparticles volume. Furthermore, polystyrene was found to significantly reduce the detrimental effects of oxidation and after 24 h of exposure in air, the polystyrene protected magnesium particles only lost 30% of their initial storage capacity without any negative effects on kinetics. Under the same conditions, the unprotected magnesium particles lost 50% of their initial hydrogen storage capacity. More remarkably, the combined effect of oxidation and polystyrene coating was found to lead to a significant reconstruction of magnesium nanoparticles upon hydrogen cycling and a drastic shift of the thermodynamics with both enthalpy an entropy significantly decreasing to 52.3 ± 3.2 kJ mol−1 H2 and 101.3 ± 4.5 J mol−1 K−1 H2, respectively. Since smaller magnesium nanoparticles were generated through this process, it is believed that the evolution of thermodynamics observed was mainly due to particle size reduction. Hence, a new tool for adjusting the thermodynamic properties of the magnesium/hydrogen reaction is through the controlled oxidation of polymer stabilised magnesium nanoparticles. Herein, this resulted in a significant decrease in enthalpy leading to weaker hydrogen bond strengths alongside a decrease in entropy resulting from the more weakly bonded and more mobile hydrogen atoms. By further unlocking this apparent interdependency between enthalpy and entropy, effectively storing hydrogen under mild conditions with magnesium may become possible.Acknowledgements
The authors gratefully acknowledge the financial support by UNSW Internal Research Grant program and under the Australian Research Council's Discovery Projects funding Scheme (project no. DP1095209). We appreciate the use of instruments in the Mark Wainwright Analytical Centre at UNSW.References
- L. Schlapbach and A. Zuttel, Nature, 2001, 414, 353–358 CrossRef CAS PubMed.
- U. Eberle, M. Felderhoff and F. Schüth, Angew. Chem., Int. Ed., 2009, 48, 6608–6630 CrossRef CAS PubMed.
- B. Sakintuna, F. Lamari-Darkrim and M. Hirscher, Int. J. Hydrogen Energy, 2007, 32, 1121–1140 CrossRef CAS PubMed.
- J. J. Vajo, S. L. Skeith and F. Mertens, J. Phys. Chem. B, 2005, 109, 3719–3722 CrossRef CAS PubMed.
- R. W. P. Wagemans, J. H. van Lenthe, P. E. de Jongh, A. J. Van Dillen and K. P. de Jong, J. Am. Chem. Soc., 2005, 127, 16675–16680 CrossRef CAS PubMed.
- V. Bérubé, G. Radtke, M. Dresselhaus and G. Chen, Int. J. Energy Res., 2007, 31, 637–663 CrossRef PubMed.
- K. C. Kim, B. Dai, J. Karl Johnson and D. S. Sholl, Nanotechnology, 2009, 20, 204001 CrossRef PubMed.
- W. Liu and K.-F. Aguey-Zinsou, J. Mater. Chem. A, 2014, 2, 9718–9726 CAS.
- O. Friedrichs, J. C. Sanchez-Lopez, C. Lopez-Cartes, M. Dornheim, T. Klassen, R. Bormann and A. Fernandez, Appl. Surf. Sci., 2006, 252, 2334–2345 CrossRef CAS PubMed.
- A. Borgschulte, M. Bielmann, A. Zuttel, G. Barkhordarian, M. Dornheim and R. Bormann, Appl. Surf. Sci., 2008, 254, 2377–2384 CrossRef CAS PubMed.
- B. J. Kooi, G. Palasantzas and J. T. M. De Hosson, Appl. Phys. Lett., 2006, 89, 161914 CrossRef PubMed.
- K. F. Aguey-Zinsou, J. Ares Fernandez, T. Klassen and R. Bormann, Mater. Res. Bull., 2006, 41, 1118–1126 CrossRef CAS PubMed.
- Z. Zhao-Karger, J. Hu, A. Roth, D. Wang, C. Kubel, W. Lohstroh and M. Fichtner, Chem. Commun., 2010, 46, 8353–8355 RSC.
- M. Paskevicius, H. Y. Tian, D. A. Sheppard, C. J. Webb, M. P. Pitt, E. M. Gray, N. M. Kirby and C. E. Buckley, J. Phys. Chem. C, 2011, 115, 1757–1766 CAS.
- T. K. Nielsen, K. Manickam, M. Hirscher, F. Besenbacher and T. R. Jensen, ACS Nano, 2009, 3, 3521–3528 CrossRef CAS PubMed.
- S. Zhang, A. F. Gross, S. L. Van Atta, M. Lopez, P. Liu, C. C. Ahn, J. J. Vajo and C. M. Jensen, Nanotechnology, 2009, 20, 204027 CrossRef PubMed.
- K.-F. Aguey-Zinsou and J.-R. Ares-Fernández, Energy Environ. Sci., 2010, 3, 526 CAS.
- K. F. Aguey-Zinsou and J. R. Ares-Fernández, Chem. Mater., 2007, 20, 376–378 CrossRef.
- K. J. Jeon, H. R. Moon, A. M. Ruminski, B. Jiang, C. Kisielowski, R. Bardhan and J. J. Urban, Nat. Mater., 2011, 10, 286–290 CrossRef CAS PubMed.
- S. Makridis, E. Gkanas, G. Panagakos, E. Kikkinides, A. Stubos, P. Wagener and S. Barcikowski, Int. J. Hydrogen Energy, 2013, 38, 11530–11535 CrossRef CAS PubMed.
- E. J. Setijadi, C. Boyer and K.-F. Aguey-Zinsou, Phys. Chem. Chem. Phys., 2012, 14, 11386–11397 RSC.
- E. J. Setijadi, C. Boyer and K.-F. Aguey-Zinsou, Int. J. Hydrogen Energy, 2013, 38, 5746–5757 CrossRef CAS PubMed.
- C. J. Orme, M. L. Stone, M. T. Benson and E. S. Peterson, Sep. Sci. Technol., 2003, 38, 3225–3238 CrossRef CAS PubMed.
- C. L. Beyler and M. M. Hirschler, SFPE handbook of fire protection engineering, 3rd edn, 2002 Search PubMed.
- K. F. Aguey-Zinsou and C. Boyer, ChemPlusChem, 2012, 77, 423–426 CrossRef CAS PubMed.
- P. Jolibois, C. R. Acad. Sci., Ser. IIa: Sci. Terre Planetes, 1912, 155, 353–355 CAS.
- E. C. Ashby, Q. Rev., Chem. Soc., 1967, 21, 259–285 RSC.
- C. Zlotea and M. Latroche, Colloids Surf., A, 2013, 439, 117–130 CrossRef CAS PubMed.
- S. J. Splinter, N. S. McIntyre, W. N. Lennard, K. Griffiths and G. Palumbo, Surf. Sci., 1993, 292, 130–144 CrossRef CAS.
- C. D. Wagner, W. M. Riggs, L. E. Davis and J. F. Moulder, Handbook of X-ray Photoelectron Spectroscopy, Perkin&Elmer Cooperation, 1997 Search PubMed.
- Z. Dehouche, J. Goyette, T. K. Bose, G. Liang, J. Huot and R. Schulz, in Electrochemical and Chemical Reactivity of Amorphous and Nanocrystalline Materials, ed. R. Schulz, 2001, vol. 377, pp. 77–83 Search PubMed.
- A. Etiemble, H. Idrissi and L. Roue, J. Power Sources, 2011, 196, 5168–5173 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: TGA/MS of PSTN, PCI curves of MgH2/C and MgH2/PSTN, and associated Kissinger plots. See DOI: 10.1039/c4ra08404g |
| This journal is © The Royal Society of Chemistry 2014 |