
Pd–Pt nanostructures on carbon nanofibers as an oxygen reduction electrocatalyst
Abstract
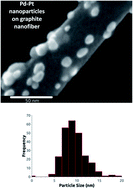
A new oxygen reduction catalyst is made of Pd and Pt nanostructures (Pd–Pt) supported on a herring-bone arrangement of carbon nanofibers (NFs) and is synthesized in one pot by the sequential reduction of Pd2+ and Pt4+ in aqueous chloride solution with ethylene glycol and then adding the carbon NF to precipitate the catalyst. The exchange current density for the oxygen reduction reaction (ORR) on Pd–Pt is 1.44 × 10−4 mA cm−2 versus 1.41 × 10−4 mA cm−2 for pure Pt as determined using a thin-film rotating disk electrode (TF-RDE) method. A single cell hydrogen anode and oxygen cathode fuel cell in which the cathode is catalyzed with Pd–Pt gives a performance as good as or better than with a commercial Pt catalyst in the cathode for the same total metal loading, 0.5 mgmetal cm−2. This shows the catalyst made of Pd–Pt supported on carbon NF is a low cost alternative to Pt on carbon, because it gives the same or better activity with less Pt.
- This article is part of the themed collection: Materials for Energy storage
 Please wait while we load your content...
Please wait while we load your content...

