
First principles investigation of point defect-related properties in Ti2AlN
Abstract
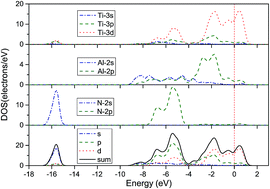
The formation and migration energies of the mono-vacancies, foreign impurities (H, He and O atoms) and interstitials in Ti2AlN have been investigated using first principles calculations. The results have shown that the mono-vacancy formation is energetically most favorable for the Al atom. There are three stable configurations for the foreign impurities, the Al-layer triangle center site, the interstitial site (⅓,⅔,0.6578) and the vacancy site. It was also found that the O substitution is easily formed. For interstitials, the octahedral interstitial (Ti, N, H or O) and Al-layer triangle interstitial (Al or He) sites were observed in our simulation. Moreover, the H, O and N interstitials are dominant in Ti2AlN. These results could provide theoretical guidance for future experiments and for the application of Ti2AlN.
 Please wait while we load your content...
Please wait while we load your content...

