
Gel-combustion synthesis and electrochemical performance of LiNi1/3Mn1/3Co1/3O2 as cathode material for lithium-ion batteries
Ying Wanga,
Hong Zhanga,
Wenhao Chena,
Zhiyuan Maa and
Zhicheng Li*ab
aSchool of Materials Science and Engineering, Central South University, Changsha 410083, P.R. China. E-mail: zhchli@csu.edu.cn; Fax: +86-731-8887-6692; Tel: +86-731-8887-7740
bState Key Laboratory of Powder Metallurgy, Central South University, Changsha 410083, P.R. China
First published on 1st August 2014
Abstract
Layered structure LiNi1/3Mn1/3Co1/3O2 (LNMC) powders were synthesized by a gel-combustion method and were calcined at 750, 800, 900 and 1000 °C, respectively. The analysis of phase composition and microstructure results show that the prepared LNMC powders have the layered structure with high order level and low cationic mixing degree. As a working electrode via Li in lithium batteries, the powders calcined at 900 °C for 5 h exhibit high electrochemical performance with an initial discharge capacity of 210 mA h g−1 at a current density of 40 mA g−1, good cycling performance and rate capacity. Detailed X-ray photoelectron spectroscopy analysis of the LNMCs, which had been induced at various charge–discharge stages, revealed that the discharge capacities of the LNMC were mainly resulted from the redox reactions of Ni2+ ↔ Ni3+ ↔ Ni4+, and were also partially attributed to the Co3+ ↔ Co4+ redox couple at electrochemical potentials above 4.1 V, while Mn4+ ions were electrochemically inactive during the charge–discharge processes.
1. Introduction
In recent years, much attention has been paid and focused on the research and development of stable energy storage devices, among which rechargeable lithium-ion batteries (LIBs) are the mainstays for their high energy density, high capacity and cycling performance.1–3 LiCoO2 is the most applied cathode material in lithium ion batteries, although it still has some disadvantages such as high cost. Mixed transition metal oxides, Li(Ni, Mn, Co)O2, have been pursued in the past decade due to their unique advantages such as lower toxicity, improved thermal stability, better cycling stability, higher reversible capacity, lower costs, etc., compared with LiCoO2.4–6 LiNi1/3Mn1/3Co1/3O2 (denoted as LNMC) with a layered structure as that of α-NaFeO2 is considered to be one of the most promising cathodes,7,8 and exhibited discharge capacities about 160–180 mA h g−1 in the cut-off range of 2.5–4.5 V.9 To improve the crystalline characteristics and electrochemical performance, several preparation methods such as solid state reaction,10 co-precipitation,11,12 spray pyrolysis,13 sol–gel method14 and combustion synthesis,15 etc., have been adopted. A traditional sol–gel method needs high calcination temperatures (1000 °C or above) and long soaking time (15–20 h),16,17 resulting in unexpected particle growth.The understanding of redox characteristics is important to modify the structure/microstructure and to improve the electrochemical performance of an LNMC. Much effort has been focused on the structural and valence changes of the delithiated LixNi1/3Co1/3Mn1/3O2 materials.18–20 Because there is no obvious structural transition during the electrochemical cycling in these compound systems,21 it is not suitable to investigate the redox mechanism by only the phase and/or structure analysis methods such as X-ray diffraction (XRD) and transmission electron microscopy (TEM), which are useful for materials with phase transition.22–25 X-ray photoelectron spectroscopy (XPS) became one of the most effective methods in studying the electrochemical characteristics of LNMC-based electrodes in LIBs,26–29 for the valence state of the transition metal cations indeed changed during the charge–discharge processes. In the reported work, XPS analysis was applied on LNMC before and/or after the full charge–discharge process. It was thus purposeful to reveal the redox characteristics by analyzing the valence states of the transition metal cations in LNMCs that were induced at different charge–discharge stages.
Herein, a sol–gel/combustion synthesis method was developed to prepare the LNMC powders, which exhibit a high electrochemical performance. Besides the microstructure characterization by XRD, scanning electron microscopy (SEM) and TEM, XPS measurements were performed to study the redox characteristics by analyzing the valence state evolution of the transition metal cations of LNMC that were charged–discharged to different oxidation–reduction states.
2. Experimental
LNMC powders were synthesized via a sol–gel/combustion method using citric acid (HOC(CH2COOH)2COOH) as the combustion agent. The amounts of Li2CO3, MnCO3, 2CoCO3·3Co(OH)2·3H2O and NiCO3·2Ni(OH)2·4H2O as the starting materials were weighed according to the ratio in the LNMC compound, and 5% excess of Li2CO3 was used to compensate for possible loss during calcination. The starting materials were dissolved in diluted nitric acid in distilled water. Then the same molar quantity of citric acid as that of all cations in the starting materials was weighed and added into the solution. The mixture was adjusted to pH value of about 8 by adding ammonia solution, and was stirred at 80 °C for 5 h to obtain a clear viscous gel. The gel was dried at 120 °C for 12 h. The xerogel was put into a crucible furnace and heated to 300 °C for combustion. The combusted porous precursor was ground and heat treated at 450 °C for 4 h in air to eliminate the organic composition. The resulted powders were pressed into pellets and calcined at 750, 800, 900 and 1000 °C, respectively, in air for 5 h. The calcined pellets were crushed and ground into powders with a mortar and pestle.The phase component of the samples were analyzed by an X-ray diffractometer (Rigaku D/MAX, 2500) with Cu-Kα radiation (λ = 0.154056 nm). The morphology and microstructure of the calcined powders were characterized by SEM (FEI Sirion 200) and TEM (FEI Tecnai G2 F20). The powder was dispersed into ethanol and a drop of the suspension was transferred onto a Cu grid with holey carbon foil and dried for TEM observations.
Li/electrolyte/LNMC 2032-type coin cells were assembled inside an argon-filled glove box. The LNMC powder, acetylene black and polyvinylidene difluoride, at a weight ratio of 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1, were mixed and dispersed in a N-methylpyrrolidone solution. The slurry was spread on an aluminum foil, and vacuum dried overnight at 80 °C. Lithium foil was used as a reference and counter electrode, polypropylene membrane (Celgard 3501) was used as a separator, and 1.0 M LiPF6 in mixed ethylene carbonate (EC) and diethyl carbonate (DEC) (the volume ratio of EC to DEC was 1:1) were employed as the electrolyte. The galvanostatic charge–discharge of the LNMC electrode was performed with a battery measurement system (Land CT2001A, China) at room temperature. The cells were tested with a voltage range of 2.5–4.5 V at 40 mA g−1. The rate capacities at various current densities (40, 80, 200 mA g−1) were also measured. Cyclic voltammetry (CV) performance was determined by an electrochemical workstation (Gamry Reference 600, USA) in the voltage range of 2.5–4.5 V at a scanning rate of 0.1 mV s−1. The electrochemical impedance spectroscopy (EIS) of LNMC was measured by the electrochemical workstation in the frequency range of 1 MHz to 1 Hz.
:1:1, were mixed and dispersed in a N-methylpyrrolidone solution. The slurry was spread on an aluminum foil, and vacuum dried overnight at 80 °C. Lithium foil was used as a reference and counter electrode, polypropylene membrane (Celgard 3501) was used as a separator, and 1.0 M LiPF6 in mixed ethylene carbonate (EC) and diethyl carbonate (DEC) (the volume ratio of EC to DEC was 1:1) were employed as the electrolyte. The galvanostatic charge–discharge of the LNMC electrode was performed with a battery measurement system (Land CT2001A, China) at room temperature. The cells were tested with a voltage range of 2.5–4.5 V at 40 mA g−1. The rate capacities at various current densities (40, 80, 200 mA g−1) were also measured. Cyclic voltammetry (CV) performance was determined by an electrochemical workstation (Gamry Reference 600, USA) in the voltage range of 2.5–4.5 V at a scanning rate of 0.1 mV s−1. The electrochemical impedance spectroscopy (EIS) of LNMC was measured by the electrochemical workstation in the frequency range of 1 MHz to 1 Hz.
XPS (K-Alpha 1063) spectra were obtained to evaluate the valence states of the as-calcined powder and the electrochemically induced LNMC. The induced materials at different charge–discharge stages for XPS analysis were selected when the cells were charged, in the 3rd cycle, at the platform (4.1 V), fully charged (4.5 V), discharged at the platform (3.8 V) and fully discharged (2.5 V) states, respectively. Then the cells were disassembled in a glove box and the electrode materials were washed out with DMC (dimethyl carbonate). The washed active materials were dispersed into acetone and dried for XPS measurements. The Thermo Avantage software was used for data analysis and peak fitting.
3. Results and discussion
3.1 Phase and microstructure characterization
The XRD patterns of the LNMC powders calcined at different temperatures are shown in Fig. 1. All the XRD patterns can be well fitted to a hexagonal layered structure with space group R3m as that of α-NaFeO2. There is no detectable diffraction peak from impurity. The diffraction peaks become stronger and sharper with the increase in calcination temperature, indicating the increase in crystallinity in the powders. The enlarged regions show that the peaks of (006)/(102) at 35–40° and (108)/(110) at 63–67° are split for the samples calcined at higher temperatures (e.g., 900 and 1000 °C). The occurrence of the split peaks of (006)/(102) and (108)/(110) indicates that a well ordered structure was formed in LNMC powders as discussed by Shi et al.30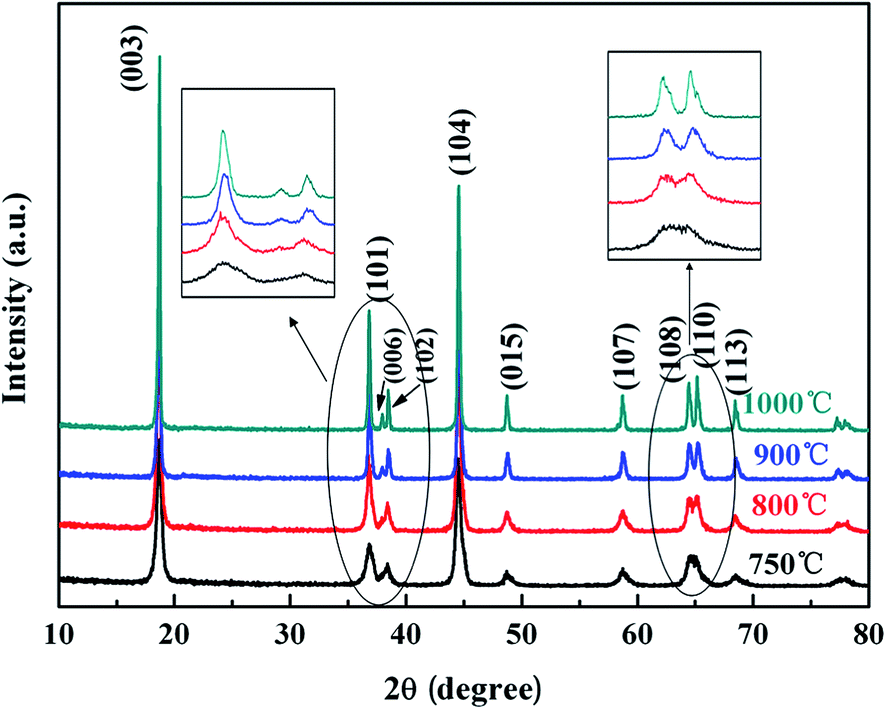 | ||
| Fig. 1 XRD patterns of LNMC powders calcined at different temperatures, showing a hexagonal layered structure with a space group of R3m as that of α-NaFeO2, and the split peaks in LNMC powders calcined at 900 and 1000 °C indicating a well ordered structure feature. | ||
The lattice parameters of the LNMC powders were calculated according to the XRD data and are listed in Table 1, where R0, Rm and c/a values were used as the essential criteria in assessing the layered LNMC crystal structure.31,32 The R0 factor defined as the ratio of (I006 + I102)/I101 is an indicator of the hexagonal ordering. A lower R0 factor is considered to be a better estimate on the separation of transition metal ions and Li+ ions onto their respective planes in a layered structure.31 The Rm factor defined as the intensity ratio of I003/I104 is an indicator of the degree of cation mixing, which strongly deteriorated the electrochemical properties of the materials. A higher value (>1.2) of Rm is desirable for a lower amount of cation mixing and better hexagonal structure. All of the c/a ratios are higher than 4.9, suggesting a well-ordered layered structure in LNMC.32 The R0 value gradually decreases with increasing calcination temperature from 750 to 900 °C, but increases only slightly at higher temperature such as 1000 °C. Similarly, maximal Rm is observed in the LNMC calcined at 900 °C. These results indicate that LNMC powders calcined at 900 °C have the best layered structure and minimal disorder level of the transition metal cations between Li (3a) and transition metal (3b) sites in the hexagonal layered structure.
| Calcination temperature (°C) | Lattice parameter | Rm I(003)/I(104) | R0[I(006) + I(102)]/I(101) | ||
|---|---|---|---|---|---|
| a (Å) | c (Å) | c/a | |||
| 750 | 2.8650 | 14.2335 | 4.9680 | 1.15 | 1.09 |
| 800 | 2.8644 | 14.2393 | 4.9708 | 1.35 | 0.75 |
| 900 | 2.8595 | 14.2197 | 4.9726 | 1.53 | 0.47 |
| 1000 | 2.8651 | 14.2412 | 4.9706 | 1.52 | 0.49 |
SEM observations of the LNMC calcined at various temperatures are shown in Fig. 2. One can see that the particles sizes are in the range of 3 to 5 μm. The inserted high magnification images show that the LNMC powers are composed of nano sized primary particles (100–500 nm). The LNMC powders calcined at 700 °C and 800 °C, seen in Fig. 2(a) and (b), have spongy-type microstructure and are comprised of irregularly agglomerated particles. The ones calcined at 900 °C and 1000 °C have highly crystalline feature (Fig. 2(c) and (d), respectively). One could also see that the primary particle sizes increased with increasing calcination temperatures.
 | ||
| Fig. 2 SEM images of LNMC powders calcined at different calcination temperatures, (a) 750 °C, (b) 800 °C, (c) 900 °C and (d) 1000 °C. | ||
Fig. 3 illustrates the TEM investigations of LNMC powders calcined at 900 °C. Fig. 3(a) shows a typical bright field TEM image of LNMC particle, which has a size of about 100–200 nm and is composed of nano-crystallites (3–6 nm). The inserted selected area electron diffraction (SAED) pattern consists of concentric rings and can be assigned to a hexagonal LNMC (space group of R3m). The SAED rings are indexed as {010}, {013}, {110}, {020} and {021} planes, respectively. Fig. 3(b) shows a high resolution TEM (HRTEM) image, providing more detailed structural information. The corresponding lattice spacings of 4.75 Å, 2.34 Å and 2.20 Å as marked in the image are in agreement with the ones of {003}, {012} and {013} in the hexagonal LNMC, respectively. Combined with the porous characteristic as shown in Fig. 2, this kind of nanostructured feature of the LNMC is convenient for Li+ to transport through the crystal lattice and related nanostructured interfaces.
 | ||
| Fig. 3 TEM investigations of LNMC materials calcined at 900 °C, showing nanostructure feature and a layered structure as that of α-NaFeO2, (a) a bright-field image and the related SAED pattern and (b) HRTEM image. | ||
3.2 Electrochemistry properties
CV was applied to explore the electrochemical reaction in the LNMC compound and typical results are shown in Fig. 4. The CV curves were obtained from the testing cell with the 900 °C-calcined LNMC as working electrode at a scan rate of 0.1 mV s−1, in the voltage range of 2.5–4.5 V, for the first three cycles. It can be seen that a pair of oxidation and reduction peaks appears at 4.10 V and 3.57 V, which could mainly be attributed to the transition metal cations in the first cycle. In the subsequent 2nd and 3rd cycles, the redox peaks shifted to 3.98 V and 3.57 V, respectively, and became almost repeatable. The nearly overlapping CV profiles in the 2nd and 3rd cycles indicate a good reversibility of lithium extraction/insertion in the LNMC. | ||
| Fig. 4 First three-cycle CV curves of an assembled cell with the 900 °C-calcined LNMC as the working electrode, at a scanning rate of 0.1 mV s−1 and ranging in 2.5–4.5 V vs. Li/Li+. | ||
Fig. 5 shows the initial charge–discharge profiles of the cells with LNMC calcined at various temperatures recorded in the range of 2.5–4.5 V at a current density of 40 mA g−1. A charge platform in the range of 3.8–4.1 V and a corresponding discharge platform in 3.9–3.6 V can be detected and could be assigned to the redox reactions of the transition metal ions. It can be seen that the initial discharge specific capacities of the LNMC calcined at 750, 800, 900 and 1000 °C are 169.6, 174.2, 210.0 and 196.7 mA h g−1, respectively. The 900 °C-calcined LNMC displays high initial charge and discharge specific capacities of 242 and 210 mA h g−1, respectively. These high charge and discharge capacities are comparable with the ones of a previously reported LNMC prepared by sol–gel method, e.g., an initial discharge specific capacity of 205 mA h g−1.33 The irreversible capacity losses in the first cycle of the LNMC calcined at 750, 800, 900 and 1000 °C are 21.3, 15.0, 13.3 and 15.4%, respectively. These irreversible capacity losses could mainly be attributed to the electrolyte decomposition and the formation of a solid electrolyte interface (SEI) on the surface of active materials during the first cycle.34
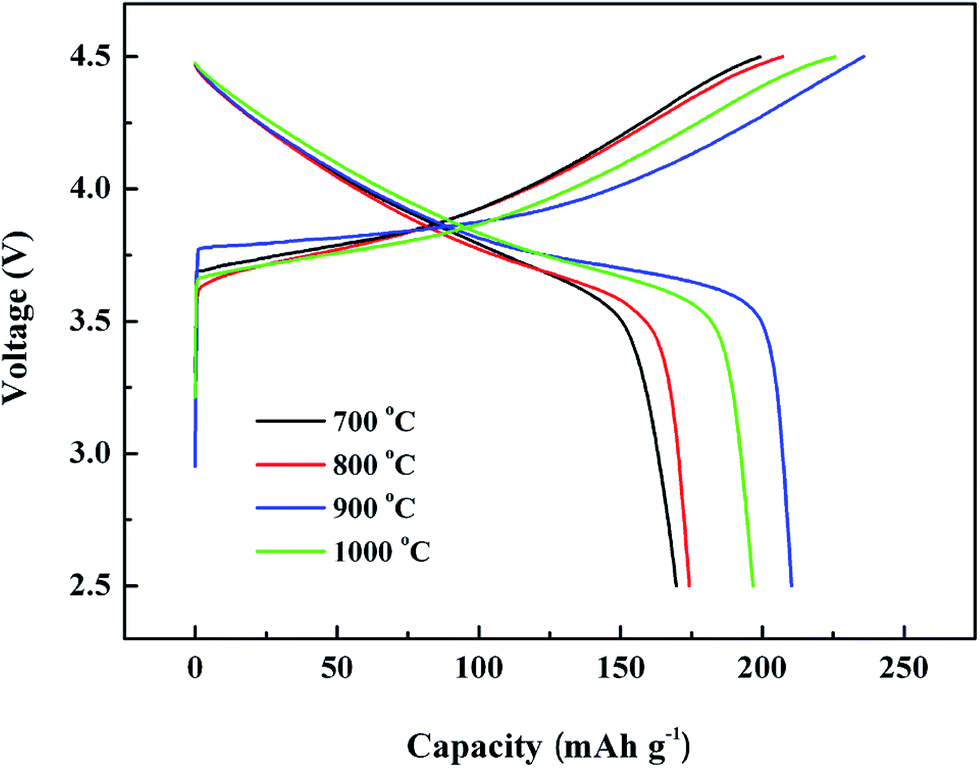 | ||
| Fig. 5 Initial charge–discharge curves of cells with the working electrode LNMC calcined at the temperatures of 750 °C, 800 °C, 900 °C and 1000 °C, respectively. | ||
The cycling performances of the LNMC calcined at different temperatures are shown in Fig. 6. It can be seen that the capacities of the LNMC electrodes decreased rapidly in the first 10 cycles. This might be attributed to the corrosion of the electrode by the electrolytes. The capacity shows a slight decrease after 10 cycles. After 50 cycles, the LNMC calcined at 750, 800, 900 and 1000 °C displayed capacities of 120, 132, 164 and 140 mA h g−1, respectively. The LNMC calcined at 900 °C exhibited the highest cycling performance and discharge capacities during the electrochemical processes in the present work.
 | ||
| Fig. 6 Comparison of cycling performances for the cells with the working electrode LNMC calcined at the temperatures of 750 °C, 800 °C, 900 °C and 1000 °C, respectively. | ||
Fig. 7 shows the rate capacities of the LNMC prepared at various calcination temperatures which were measured at different current densities. The results show an obvious difference. The 900 °C-calcined LNMC shows a high rate performance and cycling stability, e.g., discharged capacities of 195.3 mA h g−1 at 40 mA g−1, 180.1 mA h g−1 at 80 mA g−1 and 156 mA h g−1 at 200 mA g−1 (200 mA g−1 is about 1 C). When cycled back to 40 mA g−1, the corresponding capacity retention of the sample is 92.6%, compared to the initial cycle at 40 mA g−1. The rate performance is comparable to those of carbon coated LNMC, 183.5 mA h g−1 at 0.1 C, 173 mA h g−1 at 0.2 C and 155 mA h g−1 at 0.5 C.35 The high rate performance of the 900 °C-calcined LNMC can be attributed to its highly ordered layered structure and low amount of cation mixing as well as unique microstructure as discussed in Fig. 2 and 3.
 | ||
| Fig. 7 Rate capabilities of cells with the working electrodes of LNMC prepared by calcination temperatures of 750 °C, 800 °C, 900 °C and 1000 °C, respectively. | ||
It is well known that the charge–discharge efficiency is mostly dependent on the transport ability of the Li+ ions inside the active material LNMC and in the electrolyte. To investigate the transport ability of the Li+ ions in LNMC calcined at various temperatures, complex impedance spectra of the coin cells before and after 50 electrochemical cycling were collected as shown in Fig. 8. For the pristine samples, the impedance spectra in Nyquist plot are shown in Fig. 8(a). Each of the plots is composed of a depressed semicircle in the high frequency region (left part) and a straight tail in the low frequency (right part). The depressed semicircles are related to the charge transfer resistance (Rct) in the LMNC–electrolyte composite. The low-frequency tails are associated with the mass transfer resistance of Li-ion within the electrode material. The cell with 750 °C-calcined LNMC has the highest Rct while the one with 900 °C-calcined LNMC has the lowest Rct. According to the analysis on the equivalent circuit (inset in Fig. 8(a)), the values of Rct are 860.0, 550.6, 407.0 and 529.9 Ω for LNMC calcined at 750, 800, 900 and 1000 °C, respectively. These indicate that the Li+ ions have the highest transport ability in the cell of the 900 °C-calcined LNMC.
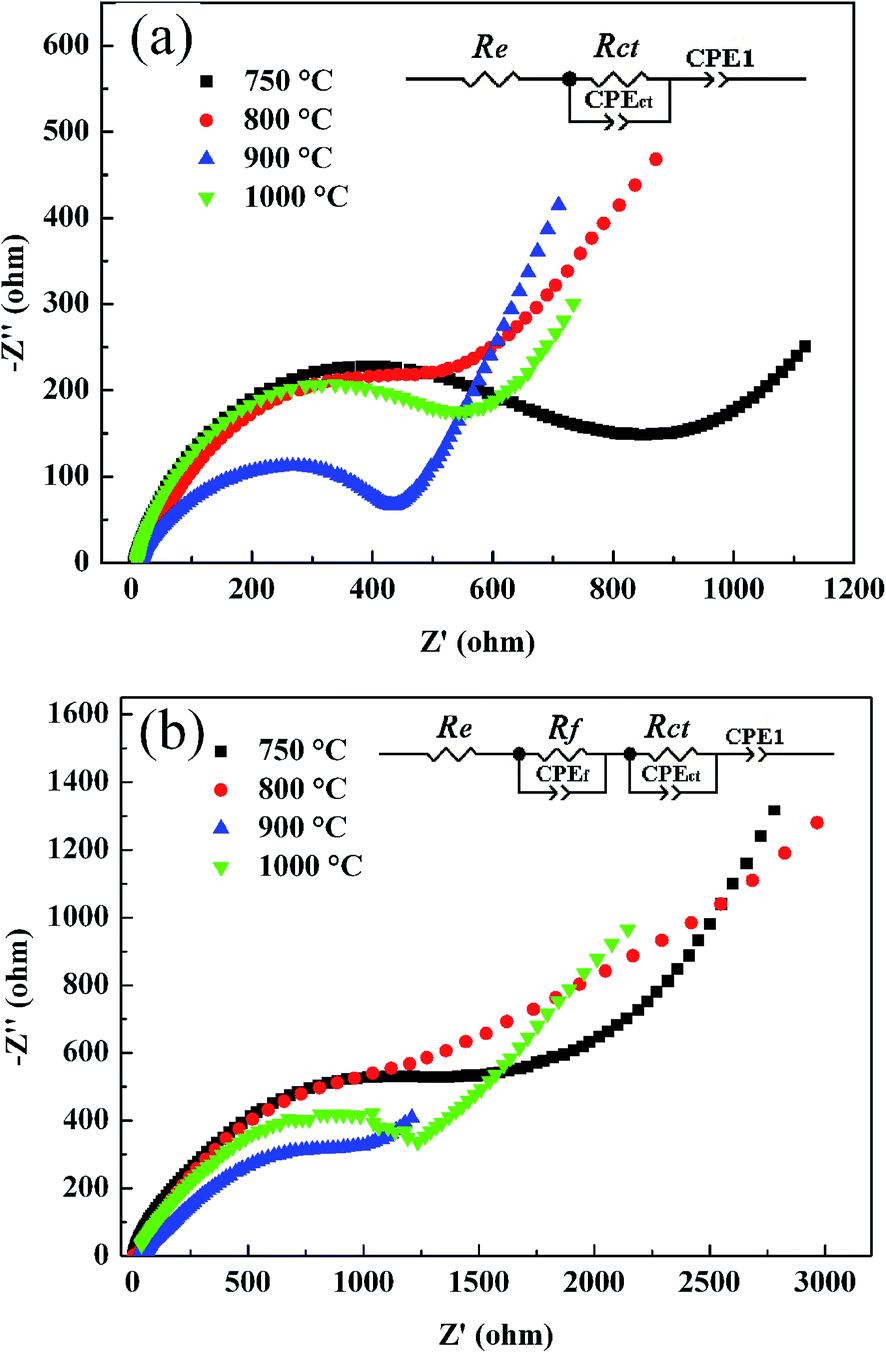 | ||
| Fig. 8 EIS spectra for the cells with LNMC calcined at different temperatures, (a) before charge–discharge measurement and (b) after 50 electrochemical cycles at 40 mA g−1. | ||
On the other hand, after 50 electrochemical cycles at a current density of 40 mA g−1, the Nyquist plots (see Fig. 8(b)) are a little different from the initial ones shown in Fig. 8(a). An additional small arc can be detected in the high frequency region, which is assigned to the impedance (Rf) of Li+ diffusion in the surface layer (SEI film). The Rf values of all cells are similar to each other and range between 50–100 Ω while the values of Rct increased distinctly for all the cells, i.e., 1643, 1528, 1037 and 1409 Ω are obtained for LNMC calcined at 750, 800, 900 and 1000 °C, respectively. The cell with 900 °C-calcined LNMC has the smallest Rct. These results infer that the 900 °C-calcined LNMC has the highest electrochemical performance.
3.3 TEM investigations of charged–discharged LNMC
To inspect the structural characteristic of LNMC during electrochemical processes, TEM investigations were performed to analyze the crystalline structure of the LNMC charged to 4.5 V and discharged to 2.5 V in the 3rd electrochemical cycle. A typical TEM observation of an LNMC charged to 4.5 V is shown in Fig. 9(a). The top-right insert of SAED pattern shows a ring-type diffraction pattern, indicating that the LNMC consists of ultra-fine crystalline grains. The SAED rings can be indexed according to the α-NaFeO2 type crystal structure to be the crystal planes of {003}, {010}, {013}, {110} and {020}, respectively. In addition, a set of HRTEM fringe with the lattice spacing of 4.92 Å in the bottom-left insert of HRTEM image in Fig. 9(a) can be detected. The lattice spacing matches the one of the {003} plane. Compared with the results discussed in Fig. 3(a), the lattice spacing of the LNMC charged to 4.5 V increased slightly. During the charge process, the unit cell volume of Li1−xNi1/3Mn1/3Co1/3O2 is almost unchanged when x < 0.65.36 During deep delithiation when x > 0.7, the Fermi level overlaps significantly with the top of the O2− 2p band and O2 escapes from the lattice.37 The lattice spacing increases with decreasing lithium content due to an increasing electrostatic repulsion across the van der Waals gap between the (Ni1/3Mn1/3Co1/3)O2 layers.20 The reaction during the charging process can be described by the followed equation.| LiNi1/3Mn1/3Co1/3O2 → Li1−xNi1/3Mn1/3Co1/3O2 + xLi+ + xe˙ | (1) |
 | ||
| Fig. 9 TEM investigations of LNMC discharged–charged to different stages in the 3rd electrochemical cycle, (a) charged to 4.5 V and (b) discharged to 2.5 V. | ||
The results confirm that the crystal structure of the 4.5 V charged LNMC remain almost the same as that of the as-prepared LNMC powder, except for the slight increase in lattice spacing.
Fig. 9(b) shows a TEM investigation of the LNMC electrode discharged to 2.5 V in the 3rd cycle. The top-right insert of diffraction rings can be assigned according to the α-NaFeO2 type LNMC structure and are indexed as {003}, {010}, {013}, {110} and {020}, respectively. The bottom-left insert in Fig. 9(b) displays the corresponding HRTEM image. A fringe spacing of 4.75 Å is inconsistent with the lattice spacing of {003} plane in Li1−xNi1/3Mn1/3Co1/3O2 charged to 4.5 V, and is in agreement with the one of the pristine powders as mentioned in Fig. 3. The results suggest that the electrode was reduced to LNMC from Li1−xNi1/3Mn1/3Co1/3O2. The TEM investigations, combined with the analysis in Sections 3.1 and 3.2, revealed that the LNMC electrode has high reversibility and stability during electrochemical cycling.
3.4 Redox mechanism analyzed by XPS
As discussed above, the LNMC compounds, especially the ones calcined at 900 °C, have good electrochemical reversibility and structure stability during the electrochemical processes. During the delithiating and lithiating reactions, the Li-ion content in LNMC changed, i.e., x changed in Li1−xNi1/3Mn1/3Co1/3O2 (x ≤ 1), and must accompanied by the reduction–oxidation reactions and the variation in the valence states of Ni, Co and/or Mn.15,26,38 XPS analysis is one of the effective ways for the studying the valence state evolution.Fig. 10 shows the 2p core level XPS spectra of Mn, Ni and Co in the LNMC active materials in pristine and different charged–discharged states. The Mn 2p3/2–Mn 2p1/2 doublets are shown in Fig. 10(a). For the pristine LNMC, the binding energies of the Mn 2p3/2 and Mn 2p1/2 are located at 642.16 eV and 653.66 eV, respectively. After being induced at various charged–discharged states, the binding energies of the Mn 2p3/2 and Mn 2p1/2 are located around the ones similar to those of the pristine sample. The slight shift of peaks should arise from the changes in the Mn local environment, instead of the changes of the oxidation state upon charge–discharge process.38 It could be inferred that the valence of Mn ions are predominantly in Mn4+ state during the electrochemical processes, confirming that the Mn4+ in LNMC is electrochemically inactive. The results are quite different than the Li-rich layered material (Li1.2Ni0.13Co0.13Mn0.54O2) in which an activated redox reaction (Mn3+/Mn4+) took place as shown by the XPS analysis by Yabuuchi et al.39
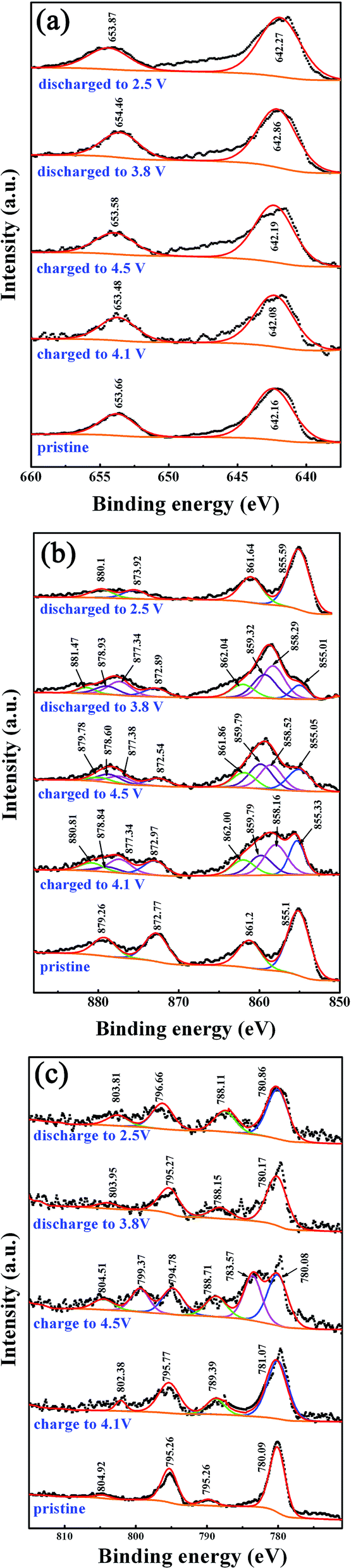 | ||
| Fig. 10 XPS analysis of the transition metal cations in the LNMC active materials electrochemically induced at various charge–discharge stages, i.e., pristine, charged to 4.1 V, charged to 4.5 V, discharged to 3.8 V and discharged to 2.5 V, (a) Mn, (b) Ni and (c) Co. | ||
The valence state evolution of the Ni cations at different electrochemical stages was analyzed by XPS as shown in Fig. 10(b). Ni 2p3/2 and Ni 2p1/2 peaks at 855.1 eV and 872.77 eV correspond to Ni2+ in the pristine LNMC. It is noted that each of the Ni 2p doublets is deconvoluted into two peaks. The small peaks located at 861.2 eV and 879.26 eV are considered to be satellite peaks. The appearance of satellite peaks could be ascribed to the multiple splitting which corresponds to the excited state in the energy bands.40 When the cell were charged to 4.1 V, besides the initial XPS peaks corresponding to Ni2+, a pair of peaks can be detected at 858.16 eV and 877.34 eV, which correspond to Ni3+. At the same time, small peaks of the binding energies at 859.79 eV and 878.84 eV corresponding to Ni4+ 2p3/2 and Ni4+ 2p1/2, respectively, can also be detected, indicating that the Ni2+ was also oxidized to Ni4+ state. This suggests that the reactions, either a one-step reaction of Ni2+ → Ni4+ or a two-step reaction of Ni2+ → Ni3+ and then Ni3+ → Ni4+, might have taken place during charge processes. When further charged to 4.5 V, the peaks corresponding to Ni4+ at 859.79 eV and 878.60 eV became stronger. Conversely, the peaks corresponding to Ni2+ and Ni3+ became weaker compared to the ones charged to 4.1 V. These altogether mean a further oxidation reaction of Ni2+/Ni3+ → Ni4+ occurred.
During the discharge process, when the LNMC was discharged to 3.8 V, the Ni3+ 2p3/2 and Ni3+ 2p1/2 peaks at 858.29 eV and 877.34 eV, respectively, are higher than those of the Ni4+ ones, indicating that the Ni4+ was partially reduced back to the Ni3+ state. It is interesting that, when LNMC was further discharged to 2.5 V, the XPS peaks corresponding to Ni3+ and Ni4+ disappeared, and the binding energies of Ni cations are almost the same as the ones of Ni2+ in pristine LNMC. This means that the full reduction of Ni3+ → Ni2+ took place. Therefore, the redox reactions of Ni2+ ↔ Ni3+ ↔ Ni4+ in LNMC could be taking place during the charge–discharge processes.
Fig. 10(c) displays the 2p core-level XPS spectra of Co ions in LNMC at the pristine and different electrochemical states. In the pristine one, the binding energy peaks at 780.09 eV and 795.26 are attributed to Co3+. The peaks at 789.78 eV and 804.92 eV can be detected and are considered to be the related satellite peaks. When charged to 4.1 V, the binding energies of Co 2p3/2 and Co 2p1/2 remain almost the same as those in the pristine LNMC. When charged to 4.5 V, a pair of extra peaks appearing at 783.57 eV and 799.37 eV can be assigned to Co4+. Please note that the peaks corresponding to Co4+ disappeared when the electrode was discharged to 3.8 V and remained absent when further discharged to 2.5 V. So it can be proposed that the redox reactions Co3+ ↔ Co4+ only took place at higher potential stages (above 4.1 V) in the LNMC materials.
Based on the discussions on the electrochemistry property, phase composition by TEM analysis, and valence states by XPS analysis, one could have a further understanding of the redox characteristics in the LNMC during the charge–discharge processes. The electrochemical mechanism in LNMC are mainly attributed to the redox reactions of Ni2+ ↔ Ni3+ ↔ Ni4+ during the whole charge–discharge processes, and the one of Co3+ ↔ Co4+ only took place at the higher potential region of above 4.1 V, while the Mn4+ is electrochemically inactive.
4. Conclusions
The layered structure of LiNi1/3Mn1/3Co1/3O2 (LNMC) powders with highly ordered level and low cation-mixing degree have been successfully synthesized via a gel-combustion method followed by 900 °C/5 h calcination. The 900 °C calcined LNMC exhibit a high discharge capacity of 210 mA h g−1 at a current density of 40 mA g−1, and high cycling performance and rate capacity. X-ray photoelectron spectroscopy analysis of the valence state evolution in the LNMC that had been induced at various charge–discharge stages is an effective method to reveal redox characteristics of the material. The electrochemical mechanism of the LNMC, induced at potentials between 2.5 and 4.5 V, is proposed to be mainly attributed to the redox reactions of Ni2+ ↔ Ni3+ ↔ Ni4+ during the whole charge–discharge processes, and the redox reactions of Co3+ ↔ Co4+ only take place at electrochemical potential region above 4.1 V, while the Mn4+ is electrochemically inactive in the whole charge–discharge processes.Acknowledgements
The authors acknowledge the support of the National Nature Science Foundation of China (no. 51172287) and the Laboratory Research Fund by the State Key Laboratory of Powder Metallurgy, China. The authors would also like to express their gratitude to Professor Weifeng Wei in the Powder Metallurgy Research Institute of Central South University for his support in LIBs assembling.References
- Z. Yang, J. Zhang, M. C. W. Kintner-Meyer, X. Lu, D. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577–3613 CrossRef CAS PubMed.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- F. X. Wang, S. Y. Xiao, Y. Y. Hou, C. L. Hu, L. L. Liu and Y. P. Wu, RSC Adv., 2013, 3, 13059–13084 RSC.
- N. Igawa, T. Taguchi, H. Fukazawa, H. Yamauchi and W. Utsumi, J. Am. Ceram. Soc., 2010, 93, 2144–2146 CrossRef CAS PubMed.
- T. F. Yi, Y. Xie, L. J. Jiang, J. Shu, C. B. Yue, A. N. Zhou and M. F. Ye, RSC Adv., 2012, 2, 3541–3547 RSC.
- H. J. Yu, Y. R. Wang, D. Asakura, E. Hosono, T. Zhang and H. S. Zhou, RSC Adv., 2012, 2, 8797–8807 RSC.
- M. S. Islam, R. A. Davies and J. D. Gale, Chem. Mater., 2003, 15, 4280–4286 CrossRef CAS.
- N. Yabuuchi and T. Ohzuku, Mater. Res. Bull., 2008, 43, 3543–3552 CrossRef PubMed.
- M. H. Lee, Y. J. Kang, S. T. Myung and Y. K. Sun, Electrochim. Acta, 2004, 50, 939–948 CrossRef CAS PubMed.
- J. M. Kim and H. T. Chung, Electrochim. Acta, 2004, 49, 937–944 CrossRef CAS PubMed.
- Y. Zhao, Y. Y. Wang, Q. Y. Lai, L. M. Chen and Y. J. Hao, Synth. Met., 2009, 159, 331–337 CrossRef CAS PubMed.
- P. Zhang, L. Zhang, X. Ren, Q. Yuan and J. Liu, Synth. Met., 2011, 161, 1092–1097 CrossRef CAS PubMed.
- S. H. Park, C. S. Yoon, S. G. Kang, H. S. Kim, S. I. Moon and Y. K. Sun, Electrochim. Acta, 2004, 49, 557–563 CrossRef CAS PubMed.
- S. Kim, S. Kim, C. Kim, W. Kim and K. Park, Mater. Lett., 2011, 65, 3313–3316 CrossRef CAS PubMed.
- G. Y. Kim, S. B. Yi, Y. J. Park and H. G. Kim, J. Power Sources, 2003, 119–121, 171–174 Search PubMed.
- M. Gozu, K. Świerczek and J. Molenda, J. Power Sources, 2009, 194, 38–44 CrossRef CAS PubMed.
- Z. D. Huang, X. M. Liu, S. W. Oh, B. Zhang, P. C. Ma and J. K. Kim, J. Mater. Chem., 2011, 21, 10777–10784 RSC.
- X. Y. Qiu, Q. C. Zhuang, Q. Q. Zhang, R. Cao, Y. H. Qiang, P. Z. Ying and S. G. Sun, J. Electroanal. Chem., 2012, 687, 35–44 CrossRef CAS PubMed.
- P. Gao, G. Yang, H. Liu, L. Wang and H. S. Zhou, Solid State Ionics, 2012, 207, 50–56 CrossRef CAS PubMed.
- J. Choi and A. Manthiram, J. Electrochem. Soc., 2005, 152, A1714–A1718 CrossRef CAS PubMed.
- M. Dahbi, J. M. Wikberg, I. Saadoune, T. Gustafsson, P. R. Svedlindh and K. Edström, Electrochim. Acta, 2009, 54, 3211–3217 CrossRef CAS PubMed.
- Y. L. Liu, H. Zhang, P. Ouyang, W. H. Chen, Y. Wang and Z. C. Li, J. Mater. Chem. A, 2014, 2, 4714–4721 CAS.
- P. Ouyang, H. Zhang, Y. L. Liu, Y. Wang and Z. C. Li, Phys. Chem. Chem. Phys., 2014, 16, 5056–5060 RSC.
- Y. L. Liu, H. Zhang, P. Ouyang, W. H. Chen and Z. C. Li, Mater. Res. Bull., 2014, 50, 95–102 CrossRef CAS PubMed.
- Y. L. Liu, H. Zhang, P. Ouyang and Z. C. Li, Electrochim. Acta, 2013, 102, 429–435 CrossRef CAS PubMed.
- J. Li, S. Xiong, Y. Liu, Z. Ju and Y. Qian, Nano Energy, 2013, 2, 1249–1260 CrossRef CAS PubMed.
- E. Shinova, R. Stoyanova, E. Zhecheva, G. F. Ortiz, P. Lavela and J. L. Tirado, Solid State Ionics, 2008, 179, 2198–2208 CrossRef CAS PubMed.
- S. Leroy, H. Martinez and R. Dedryvère, Appl. Surf. Sci., 2007, 253, 4895–4905 CrossRef CAS PubMed.
- X. Zuo, C. Fan, J. Liu, X. Xiao, J. Wu and J. Nan, J. Power Sources, 2013, 229, 308–312 CrossRef CAS PubMed.
- S. J. Shi, J. P. Tu, Y. J. Mai, Y. Q. Zhang, Y. Y. Tang and X. L. Wang, Electrochim. Acta, 2012, 83, 105–112 CrossRef CAS PubMed.
- X. Zhang, A. Mauger, Q. Lu, H. Groult, L. Perrigaud, F. Gendron and C. M. Julien, Electrochim. Acta, 2010, 55, 6440–6449 CrossRef CAS PubMed.
- S. K. Martha, H. Sclar, Z. S. Framowitz, D. Kovacheva, N. Saliyski, Y. Gofer, P. Sharon, E. Golik, B. Markovsky and D. Aurbach, J. Power Sources, 2009, 189, 248–255 CrossRef CAS PubMed.
- J. Xu, S. L. Chou, Q. F. Gu, H. K. Liu and S. X. Dou, J. Power Sources, 2013, 225, 172–178 CrossRef CAS PubMed.
- Z. Wang, Y. Sun, L. Chen and X. Huang, J. Electrochem. Soc., 2004, 151, A914–A921 CrossRef CAS PubMed.
- C. T. Hsieh, C. Y. Mo, Y. F. Chen and Y. J. Chung, Electrochim. Acta, 2013, 106, 525–533 CrossRef CAS PubMed.
- J. Li, Z. R. Zhang, X. J. Guo and Y. Yang, Solid State Ionics, 2006, 177, 1509–1516 CrossRef CAS PubMed.
- H. Kobayashi, Y. Arachi, S. Emura, H. Kageyama, K. Tatsumi and T. Kamiyama, J. Power Sources, 2005, 146, 640–644 CrossRef CAS PubMed.
- Y. M. Tsai, B. J. Hwang, G. Ceder, H. S. Sheu, D. G. Liu and J. F. Lee, Chem. Mater., 2005, 17, 3191–3199 CrossRef CAS.
- N. Yabuuchi, K. Yoshii, S. T. Myung, I. Nakai and S. Komaba, J. Am. Chem. Soc., 2011, 133, 4404–4419 CrossRef CAS PubMed.
- F. Wu, M. Wang, Y. Su and S. Chen, J. Power Sources, 2009, 189, 743–747 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |