DOI:
10.1039/C4RA06196A
(Paper)
RSC Adv., 2014,
4, 35116-35123
Selective detection of hydrogen peroxide vapours using azo dyes
Received
24th June 2014
, Accepted 28th July 2014
First published on 29th July 2014
Abstract
A rapid and selective visual colour method is described for detection of hydrogen peroxide (H2O2) and peroxide based explosive (PBE) vapours by the combination of three azo dyes – Calmagite, Orange G and Orange II. The bleaching of these dyes by H2O2 is catalysed by MnII. At pH 8.0 (EPPS, N-2-hydroxyethyl-piperazine-N′-3-propanesulfonic acid) Calmagite is quickly degraded but under these conditions Orange G and Orange II are not perceptibly bleached, especially in the presence of ethylenediaminetetraacetic acid (H4edta). However, fast bleaching of Orange G and Orange II was observed at pH 9.0 (Na2CO3, sodium carbonate) due to the in situ formation of the carbonate radical (CO3−˙). Hence by a combination of Calmagite at pH 8.0, and Orange G and Orange II at pH 9.0, selectivity to H2O2 vapours against Cl2, NO2 and O3 can be demonstrated. Initial studies were carried out on filter papers but reaction times were slow. With Calmagite rapid colour changes, within 15 minutes, were found when the dye was deposited on to polyvinyl alcohol (PVA) polymer which had been coated onto a borosilicate glass plate. However, with Orange G and Orange II the colour changes on the PVA/plates were slow and this may be due to the limited availability of CO2, as the activating species, in generating CO3−˙.
Introduction
Peroxide based explosives (PBEs) are synthesised from hydrogen peroxide (H2O2) and contain peroxide linkages incorporated within a cyclic carbon structure such that the scaffolding holds the reacting atoms in close proximity to each other. This is shown in the structures (Fig. 1) of triacetone triperoxide (TATP) and hexamethylene triperoxide diamine (HMTD).
 |
| | Fig. 1 The structures of TATP and HMTD. | |
The explosive nature of PBEs is associated with the weakness of peroxide bonds (ΔHbond (O–O) = 147 kJ mol−1) giving spin-allowed reactions under ambient conditions hence, unlike reactions of O2, activation energies are low, and the heat generated in producing large volumes of thermodynamically stable gaseous products (CO2, H2O, acetone etc.).1,2 The explosive power of TATP has been described as an “entropic burst” but the negative enthalpy of this reaction cannot also be ignored which together generate large amounts of free energy that leads to rapid acceleration in reaction rates resulting in an explosive power comparable to that of TNT.3
It is reported that PBEs were first used in 19804 but have gained notoriety in their more recent uses including the Casablanca explosions in 2003,5 the London public transportation attacks in 20056 and the transatlantic flight bombing attempt in 2006.5 These events draw attention to the need for reliable and selective detection methods for PBEs. The ready availability of the simple chemicals required to manufacture PBEs and the relative ease of their syntheses allow for the clandestine manufacture of such explosives using “kitchen sink” methodologies. For example, H2O2 is used in hair and teeth bleaching, cleaning products and disinfectants, sulfuric acid is used as a household drain cleaner and acetone as nail varnish remover. Indeed there are concerns that TATP can be assembled from these innocuous precursor chemicals at the target site.
As well as their ease of manufacture, PBEs are very difficult to detect because they lack a nitro (NO2) group which by contrast makes nitro-based explosives (NBEs) easy to detect by mass spectrometry, and they also lack absorbance in the UV. Several detection methods for PBEs and H2O2 have been described in a review by Burks and Hage.4 These include colorimetric, spectroscopic (IR, Raman, luminescence, fluorescence) and electrochemical methods. However, these techniques are more suited to laboratories and the authors emphasised the need for portable or handheld devices such as test strips for the field detection of PBEs and their precursors. PBEs are volatile and generate a high vapour pressure7 under ambient conditions and therefore detection methods can be developed to detect H2O2 in the vapour, with the use of trained sniffer dogs being reported.8 Simple portable devices that are selective and sensitive to H2O2 vapour would be ideal for the rapid detection of PBEs at public places like airports and train stations.
Progress in the development of rapid colorimetric methods for the detection of H2O2 vapours have been made by Mills9 and Suslick.10 Mills used a single green/blue azo dye Lissamine Green (LG) dissolved in polyvinyl alcohol (PVA) which was rapidly bleached in the presence of H2O2 vapours. However, chlorine (Cl2), nitrogen dioxide (NO2) and ozone (O3) also produced bleaching of LG indicating non-selectivity to H2O2. By using a combination of sixteen dyes in an array, Suslick was able to show selectivity to H2O2 vapours with detection limits below 2 ppb. However, a flatbed scanner is needed to analyse the array and the need for so many dye formulations is also undesirable.
The bleaching of the azo dyes Calmagite, Orange G and Orange II using in situ generated H2O2 has been described.11 These results show that at pH 8.0, the o,o-dihydroxy dye Calmagite is rapidly bleached but the monohydroxy azo dyes Orange II and Orange G (Fig. 2) are not bleached, or their reactions are very slow.
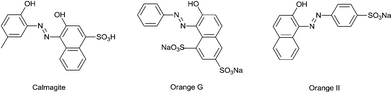 |
| | Fig. 2 The structures of Calmagite, Orange G and Orange II. | |
When using added H2O2, Orange G and Orange II are rapidly bleached at pH 9.0 in the presence of added sodium carbonate (Na2CO3). It has been suggested that this is due to the in situ generated peroxycarbonate ion (HCO4−), a more potent oxidising agent,12–14 formed (eqn (1)) from the reaction of H2O2 with bicarbonate (HCO3−).
| | |
H2O2 + HCO3− → HCO4− + H2O
| (1) |
However, Fridovich15 argues that it is CO2 and not HCO3− that is the enhancing species and that it is the carbonate radical (CO3−˙) not HCO4− that is responsible for the oxidative degradation of the dye. This work provides some evidence to support the involvement of CO2/CO3−˙. In any case, it should be possible to selectively detect H2O2 vapours from a suitable combination of the bleaching of Calmagite at pH 8.0 (EPPS), and Orange G and Orange II at pH 9.0 (Na2CO3), with a lack of bleaching of Orange G and Orange II at pH 8.0 (EPPS), especially in the presence of the complexing agent ethylenediaminetetraacetic acid (H4edta). In the case of Cl2, NO2 and O3, no discrimination would be expected thus providing selectivity to H2O2 and PBE vapours. These colour tests can be used in combination with the use of aqueous, acidified, titanium(IV) which is known to form a bright yellow-orange coloured complex,16, [Ti(O2)(OH)(H2O)3]+ with solutions containing H2O2 (eqn (2)) due to a ligand-to-metal charge transfer (LMCT).17
| | |
[Ti(OH)3(H2O)3]+ + H2O2 → [Ti(O2)(OH)(H2O)3]+ + 2H2O
| (2) |
Experimental
Materials
All the reagents used were purchased from Sigma-Aldrich. The polymer used to make the ink films was poly(vinyl alcohol) [PVA, Mw 146![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–186000, 99+% hydrolysed]. Square (22 mm) borosilicate glass plates were used to provide a transparent substrate for the inks. De-ionised water (ELGA Purelab) was used in all experiments. Filter papers were purchased from Whatman (No. 1). Hydrogen peroxide solutions were prepared from the dilution of 30% (w/w) aqueous H2O2 stock solutions. Aqueous titanium(IV) in sulfuric acid (10%) was prepared according to reported methods.18 Nitrogen dioxide was generated by the dropwise addition of concentrated HNO3 onto copper turnings while chlorine gases was generated by the dropwise addition of concentrated HCl on to MnO2; ozone was generated via an O2 cylinder using a Wallace & Tiernan Type BA.023 laboratory ozonator with the voltage set at 200 V and with O2 gas at a flow rate of 150 L h−1. All three gases were separately fed into desiccators containing filter papers or ink coated plates.
000–186000, 99+% hydrolysed]. Square (22 mm) borosilicate glass plates were used to provide a transparent substrate for the inks. De-ionised water (ELGA Purelab) was used in all experiments. Filter papers were purchased from Whatman (No. 1). Hydrogen peroxide solutions were prepared from the dilution of 30% (w/w) aqueous H2O2 stock solutions. Aqueous titanium(IV) in sulfuric acid (10%) was prepared according to reported methods.18 Nitrogen dioxide was generated by the dropwise addition of concentrated HNO3 onto copper turnings while chlorine gases was generated by the dropwise addition of concentrated HCl on to MnO2; ozone was generated via an O2 cylinder using a Wallace & Tiernan Type BA.023 laboratory ozonator with the voltage set at 200 V and with O2 gas at a flow rate of 150 L h−1. All three gases were separately fed into desiccators containing filter papers or ink coated plates.
Preparation of inks and plates
PVA crystals (10 g) were dissolved in water (90 mL) at 90 °C, cooled to room temperature and stirred overnight. For filter paper studies the dye inks were prepared by making up 10.0 mM aqueous solutions of the different dyes and then mixing aliquots (4.00 mL) with either aqueous manganese(II) chloride tetrahydrate (MnCl2·4H2O, 5.00 mM, 1.00 mL) or ethylene diamine tetraacetic acid (H4edta, 1.00 mM, taken to pH 7 using aqueous NaOH, 1.00 mL), EPPS (N-2-hydroxyethyl-piperazine-N′-3-propanesulfonic acid, 0.50 M, taken to pH 8.0 using aqueous NaOH, 1.00 mL) or sodium carbonate (0.50 M, taken to pH 9.0 using aqueous HCl, 1.00 mL) and ethanol (1.00 mL). Dye solutions were spotted on to filter papers which were then dried in a desiccator before use. For ink films, the above procedure was repeated except that 0.10 mmol of solid dye was used e.g. for Calmagite 0.0358 g of the dye was used. The PVA and inks were then cast onto the glass plates using a spin coater. A few drops of the PVA solution were deposited on to the surface of the plate which was then spun at 2400 rpm for 25 s, then a few drops of the ink solution were added onto the PVA layer and the plate spun again at 2400 rpm for 25 s. The substrate was dried using a stream of hot air from a hair dryer for 30 s and allowed to cool to room temperature before use. The dye/PVA plates were stored in a desiccator under ambient conditions.
The methodology adopted of coating PVA first on to glass plates and then the dye inks onto PVA was adopted because when solid Calmagite was added to the PVA solution (using the method by Mills9), the ink would solidify immediately which made application of the ink to the glass plates very difficult. As soon as both layers were applied they mixed naturally. Dissolving the dye in aqueous solution first and then mixing it with the PVA also worked. All three methods were tried and gave comparable results. Keeping the dye solution and PVA separate meant a much longer shelf life for the ink components.
UV-visible spectrometry
Spectroscopic measurements were made using a Jenway 7315 scanning spectrophotometer with the filter papers or plates attached to the cuvette holder using blue tac. For kinetic measurements a 1 cm plastic cuvette was used into which were drilled two 2 mm holes at a vertical separation of 3 mm along the centre of the cell and ∼10 mm above the base. The base of the cell was filled with aqueous H2O2 and the PVA/dye plates were attached to the cuvette holder facing inwards so that they could be exposed to the H2O2 vapours exiting the cuvette through the drilled holes. The cuvette was capped and a tight seal all the way around the plate and the cuvette holder was achieved using a flat layer of blue tac attached to the holder. In this way all the H2O2 vapours would be exposed only to the plate. Before each experiment, the aqueous H2O2 in the well of the cuvette was allowed to equilibrate with its headspace. Under these conditions the vapour pressure of the H2O2, pH2O2, is directly proportional to the concentration of H2O2 in solution.19
Results and discussion
Bleaching of azo dyes in aqueous solution
It has been previously11 shown that Calmagite is rapidly bleached at pH 8.0 (EPPS, N-2-hydroxyethylpiperazine-N′-3-propanesulphonic acid, 50.0 mM) by in situ generated H2O2 from the manganese(II) catalysed reduction of O2. However, under these conditions, neither Orange G (O G) nor Orange II (O II) is bleached rapidly. These results can be repeated using added H2O2. Fig. 3 (left) shows the dark purple colour of an aqueous solution of Calmagite (0.100 mM) at pH 8.0 (EPPS) and the disappearance of the colour after ∼30 min in the presence of added H2O2 (50.0 mM) and aqueous Mn2+ (5.00 μM) as catalyst. Fig. 3 (right) shows that while both O G and O II are bleached at pH 9.0 (Na2CO3), neither are perceptibly bleached at pH 8.0 (EPPS).
 |
| | Fig. 3 (left) Aqueous Calmagite (0.100 mM) at pH 8.0 (EPPS, N-2-hydroxyethylpiperazine-N′-3-propanesulphonic acid, 50 mM) before (purple) and after (pale yellow) addition of H2O2 (50.0 mM) and aqueous Mn2+ (5.00 μM). (right) Aqueous Orange G and Orange II at pH 8.0 (EPPS, N-2-hydroxyethylpiperazine-N′-3-propanesulphonic acid, 50 mM), and Orange G and Orange II at pH 9.0 (sodium carbonate, 50 mM) after the addition of H2O2 (50.0 mM) and aqueous Mn2+ (5.00 μM). | |
This would form the basis for the selective detection of H2O2 vapours as the rationale is that only in the presence of H2O2 can HCO4− or CO3−˙ be generated in situ and this is required for the oxidative degradation of O G and O II. It should also be noted that the rate of bleaching of O G and O II in the presence of sodium carbonate (Na2CO3) is pH dependent, with the rate of bleaching at pH 8.0 being very slow.
Visual filter paper tests using vapours
Fig. 4 shows the colours of filter papers coated with Calmagite at pH 8.0 (dark purple), O G at pH 8.0 and pH 9.0 (yellow-orange), O II at pH 8.0 and pH 9.0 (dark orange) and Ti(IV) (colourless) before exposure to any vapours.
 |
| | Fig. 4 Filter papers coated with azo dyes or aqueous Ti(IV) before exposure to H2O2 vapours. From top (clockwise): Calmagite at pH 8.0, Orange G at pH 8.0, Orange II at pH 8.0, Orange II at pH 9.0, Orange G at pH 9.0. Centre: Ti(IV). | |
After exposure to H2O2 vapours from solutions containing 0.50 M or 5.0 M H2O2 for 24 h in shallow petri dishes (Fig. 5) under ambient conditions, the Calmagite disks were almost completely bleached (purple to pale pink), the O G pH 9.0 disks were completely bleached (yellow-orange to colourless) and the O II pH 9.0 disks were partially bleached (dark orange to light orange), with the 5.0 M H2O2 vapour disk exhibiting more bleaching than the one exposed to 0.50 M H2O2. In contrast, the O G and O II disks at pH 8.0 showed no bleaching.
 |
| | Fig. 5 Filter papers coated with azo dyes or aqueous Ti(IV) after exposure to 0.50 M (left) or 5.0 M (right) H2O2 vapours. From top (clockwise): Calmagite at pH 8.0, Orange G at pH 8.0, Orange II at pH 8.0, Orange II at pH 9.0, Orange G at pH 9.0. Centre: Ti(IV). | |
Control experiments with pure water rather than aqueous H2O2 showed no bleaching of any of the disks indicating that bleaching was due to H2O2 vapours and not degradation of the dye through environmental exposure. These results are consistent with the aqueous solution studies and indicate pH selective bleaching of the O G and O II azo dyes which in combination with the bleaching of Calmagite and the formation of a yellow colour (Fig. 5, centre) of [Ti(O2)(OH)(H2O)3]+ peroxo complex with Ti(IV) provide a potentially useful way to distinguish H2O2 vapours from other chemical vapours.
Fig. 6 shows the effect of exposure of the filter papers to Cl2(g), NO2(g)and O3(g) for 24 h under ambient conditions.
 |
| | Fig. 6 Filter papers coated azo dyes or aqueous Ti(IV) before exposure (a) or after exposure to (b) Cl2(g), (c) NO2(g) and (d) O3(g). From left to right (in each row): Orange G at pH 8.0/Orange G at pH 9.0, Orange II at pH 8.0, Orange II at pH 9.0 and Ti(IV). Bottom row: Calmagite at pH 8.0. | |
It is immediately clear the bleaching results are very different to those with H2O2 vapours. With Cl2(g) all the azo dye disks are fully bleached, with the exception of O II at pH 8.0 and pH 9.0 which are ∼95% bleached and no reaction with Ti(IV) is discernible. In the case of NO2(g), all the azo dye disks are partially bleached with the exception of O II at pH 8.0 where there appears to be only partial bleaching and again there appears to be no reaction with Ti(IV). Lastly, with O3(g) there is only partial bleaching of the azo dyes (with the change of Calmagite being the most distinctive) and with the Ti(IV) disk there is a definite yellowing which could in isolation give a false positive for H2O2 vapours. This may be caused by the formation of a Ti(IV)–O3 complex which is also yellow or the catalysed reduction of O3 to H2O2, with the subsequent formation of the Ti(IV)-peroxo complex (eqn (2)). These somewhat surprising results with O3 were reproducible.
The sensitivity of Ti(IV) to aqueous H2O2 and H2O2 vapours is shown in Fig. 7 where concentrations as low as 0.010 M (∼3 ppm) can be detected and may be used to estimate the concentration of H2O2 vapour.
 |
| | Fig. 7 Filter paper (left) soaked with aqueous Ti(IV) and then dried with the addition of either deionised water (‘0.0’) or different aqueous [H2O2] (‘0.01’ to ‘0.25’ M) or filter paper discs (right) soaked with Ti(IV) and then dried and placed over sample vials containing either deionised water (‘H2O only’) or different aqueous [H2O2] (‘0.01’ to ‘0.25’ M). | |
Visual film tests using vapours
In general faster response times were obtained when the dye and Ti(IV) substrates were coated as ink films onto PVA coated borosilicates plates that were placed on top of vials containing 5.0 M (∼15%) solutions of H2O2 (Fig. 8). Calmagite at pH 8.0 is visibly bleached within 15 min (indeed changes are observed within the first 5 min). This increase in reactivity can be attributed to a positive change in the microenvironment polarity when the dye is encapsulated in a PVA film.9 However, the changes in colour for O G and O II on PVA are not similarly quickened. At pH 9.0 O G appears to go a mottled or speckled orange, which gives an indication of a reaction before bleaching completely within 24 h. In contrast O G at pH 8.0 shows no change over a 24 h period. The colour changes for O II are less visually clear but there is some bleaching between 15 min and the end of the test period at pH 9.0 while at pH 8.0 there is no change at 15 min. The Ti(IV) plate showed a clear yellowing of the film with within 15 min, indicating the presence of H2O2 vapours.
 |
| | Fig. 8 Images of films after exposure to vapours from 5.0 M (∼15%) aqueous solutions of H2O2. | |
These experiments were repeated using Cl2(g), NO2(g) and O3(g) (Fig. 9).
 |
| | Fig. 9 Images of films (a) before and after exposure to vapours from (b) Cl2(g), (c) NO2(g) and (d) O3(g). | |
Again it is clear that results with these three gases are very different to those of H2O2. The most dramatic dye bleaching is observed with NO2(g), with no change in the Ti(IV) film. Most surprisingly, O3(g) shows virtually no bleaching of the azo dyes but there is a colourisation of the Ti(IV) plate suggesting that the yellowing of Ti(IV) is not selective to H2O2. In the case of Cl2(g) rapid bleaching of Calmagite and O G at pH 8.0 is observed, while the bleaching of the other azo dyes is much slower, with no change in colour of the Ti(IV) plate.
Spectral studies using vapours
To enable a quantitative basis for visual studies and to enable the possible use of electronic devices to be combined with the changes in the colour of films, visible spectra (nominally 400–800 nm) were recorded before, after and, if appropriate, during the changes occurring on the films. These spectra are displayed in Fig. 10–15 and confirm the visual results from both the filter paper and film tests – with the exception of the bleaching of O II at pH 9.0 (Fig. 14) where little change in absorbance over a period of 90 minutes was observed. Fig. 16 shows that oxidative degradation of O II at pH 9.0 as measured by its change in absorbance at 490 nm does occur on the film but the reaction is very slow with a change of only ∼0.04 absorbance units over this period. This spectral data supports the visual tests on the O II pH 9.0 film. It is clear, as stated earlier, that for this particular substrate (and perhaps to a lesser extent O G), the PVA/plate appears to have an inhibitory effect on the reaction of H2O2 vapours with the dye substrates.
 |
| | Fig. 10 The absorption spectrum of a Calmagite film at pH 8.0 before (blue) and after (red) exposure to vapours for 10 min from a 5.0 M (∼15%) aqueous solution of H2O2. | |
 |
| | Fig. 11 The absorption spectrum of a Orange G film at pH 8.0 during exposure to vapours at 10 min intervals from a 5.0 M (∼15%) aqueous solution of H2O2. | |
 |
| | Fig. 12 The absorption spectrum of an Orange G film at pH 9.0 during exposure to vapours at 10 min intervals from a 5.0 M (∼15%) aqueous solution of H2O2. | |
 |
| | Fig. 13 The absorption spectrum of an Orange II film at pH 8.0 during exposure to vapours at 10 min intervals from a 5.0 M (∼15%) aqueous solution of H2O2. | |
 |
| | Fig. 14 The absorption spectrum of an Orange II film at pH 9.0 during exposure to vapours at 10 min intervals from a 5.0 M (∼15%) aqueous solution of H2O2. | |
 |
| | Fig. 15 Normalised absorption spectrum of a Ti(IV) film before (blue) and after (red) exposure to vapours from a 5.0 M (∼15%) aqueous solution of H2O2. | |
 |
| | Fig. 16 A plot of ΔAbs490 against time for the Orange II film at pH 9.0 during exposure to vapours from a 5.0 M (∼15%) aqueous solution of H2O2. | |
The most rapid (within 5 min) and distinctive bleaching is with the dark purple Calmagite dye and this in itself provides a rapid (but non-selective) indication of the presence of oxidising vapours.
Mechanisms of dye bleaching
The bleaching of Calmagite at pH 8.0 (EPPS) is fast and requires less aggressive oxidative conditions than O G and O II with carbonate not being required to achieve oxidative degradation. This may be related to the greater coordinating properties of Calmagite that has a o,o-dihydroxy motif enabling it to act as a tridentate ligand to a metal centre, compared to O G and O II that can only bind in a bidentate fashion. A mechanism for Calmagite bleaching can be proposed of the attack of coordinated of hydroperoxo ion (O2H−) on coordinated Calmagite (eqn (3)–(5) and Scheme 1).| | |
MnII + H3CAL ⇌ [MnII(CAL)]− + 3H+
| (3) |
| | |
[MnII(CAL)]− + 1½H2O2 → [MnIII(CAL)(O2H)]− + H2O
| (4) |
| | |
[MnIII(CAL)(O2H)]− → oxidised CAL → CO2 + H2O + N2
| (5) |
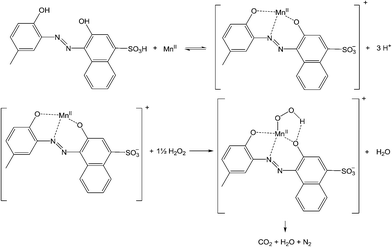 |
| | Scheme 1 The manganese catalysed oxidative degradation of Calmagite by H2O2 vapours. | |
This is similar to the mechanism proposed for the oxidative degradation of Calmagite using in situ generated H2O2, where EPR solution studies suggested the presence of MnIII but no MnIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species.11 Under these conditions (pH 8.0 and in the absence of sodium carbonate), O G and O II are bleached only very slowly, presumably because they only weakly coordinate to MnII. At pH 9.0 (Na2CO3), rapid bleaching of O G and O II was observed in solution and on filter papers but the reactions were more sluggish with the inks coated on to PVA coated borosilicate glass plates. The oxidative degradation of O II by peracetic acid catalysed by the oxo-bridged [Mn2III/IV(μ-O)2(bpy)4](ClO4)3 complex and its mononuclear analogue [MnII(bpy)2Cl2] have been studied by van Eldik et al. who proposed that the reaction proceeds via the in situ formation of HCO4−/HOOCO2− which on coordination to MnII results in the formation of MnIVO (Scheme 2).14 In this proposed mechanism there is a requirement for at least weak binding of O II to the manganese centre to effect its oxidative degradation. Yin has proposed a different mechanism for the HCO3− activated H2O2 oxidative degradation of methylene blue using supported CoII that involves the carbonate radical (CO3−˙) and possibly singlet dioxygen (1O2).20 The involvement of the CO3−˙ is also supported by Fridovich in which the terminal step would involve a one-electron oxidation of the organic substrate (SH) leading to its oxidative decomposition (eqn (6)).15
O species.11 Under these conditions (pH 8.0 and in the absence of sodium carbonate), O G and O II are bleached only very slowly, presumably because they only weakly coordinate to MnII. At pH 9.0 (Na2CO3), rapid bleaching of O G and O II was observed in solution and on filter papers but the reactions were more sluggish with the inks coated on to PVA coated borosilicate glass plates. The oxidative degradation of O II by peracetic acid catalysed by the oxo-bridged [Mn2III/IV(μ-O)2(bpy)4](ClO4)3 complex and its mononuclear analogue [MnII(bpy)2Cl2] have been studied by van Eldik et al. who proposed that the reaction proceeds via the in situ formation of HCO4−/HOOCO2− which on coordination to MnII results in the formation of MnIVO (Scheme 2).14 In this proposed mechanism there is a requirement for at least weak binding of O II to the manganese centre to effect its oxidative degradation. Yin has proposed a different mechanism for the HCO3− activated H2O2 oxidative degradation of methylene blue using supported CoII that involves the carbonate radical (CO3−˙) and possibly singlet dioxygen (1O2).20 The involvement of the CO3−˙ is also supported by Fridovich in which the terminal step would involve a one-electron oxidation of the organic substrate (SH) leading to its oxidative decomposition (eqn (6)).15
| | |
CO3−˙ + SH → HCO3− + S˙(→ CO2 + H2O + N2)
| (6) |
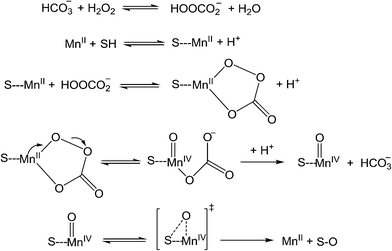 |
| | Scheme 2 Mechanism proposed by van Eldik et al. for the MnII catalysed oxidative degradation of Orange II (SH) by H2O2 in the presence of carbonate buffer at pH 8–9. | |
While the formation of CO3−˙ can be thought to be easily generated from the one-electron oxidation of HCO3− coordinated to MnIII or MnIV species, Fridovich intriguingly suggests that the presence of CO2 may enhance the MnII catalysed peroxidation of a substrates by the following mechanism (eqn (7)–(10)).
| | |
MnII + H2O2 ⇌ [MnIV(O)]2+ + H2O
| (7) |
| |
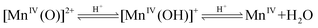 | (8) |
| | |
MnIV + CO2 ⇌ MnIII + CO2+
| (9) |
| | |
CO2+ + HO− ⇌ CO3−˙ + H+
| (10) |
It can be argued that the presence of CO2 displaces the equilibrium of reaction (7) to the right by consuming [MnIV(O)]2+ and this enhances the rates of consumption of both MnII and H2O2 and regenerates MnIII. While it may seem strange that CO2 is more readily oxidised than HCO3−, Fridovich argues that as well as an uncharged linear molecule, an alternative resonance form that can exist for CO2 is one with one oxygen atom positive and the other negative (Scheme 3).21
 |
| | Scheme 3 Resonance forms for CO2. | |
In this case the loss of an electron from the negative oxygen atom yields CO2+ and this may explain why CO2 is more readily oxidised than HCO3−. This result would explain the sluggish bleaching of O G and especially O II inks deposited on an impermeable PVA/borosilicate plate, which is somewhat off-set by the favourable polar environment created by the PVA, while on porous filter paper the reactions are slow but not inhibited.
Conclusions
The colour test results from exposure to H2O2 vapours are distinctive from the results obtained with Cl2, NO2 and O3 giving general selectivity to H2O2. While the formation of [Ti(O2)(OH)(H2O)3]+ with Ti(IV) provides a sensitive colour test for H2O2 vapours it cannot be used in isolation since O3(g) also produces a yellowing of the Ti(IV) film. The difference in reaction of H2O2 vapours to O G and O II at pH 8.0 compared to that at pH 9.0 on filter papers provide for a selective method for the detection of H2O2 vapours. As such this work provides an advance on the system described by Mills et al. using LG where no discrimination between H2O2, Cl2, NO2 and O3 was reported.9 Indeed the use of Ti(IV) with just O G at pH 8.0 and pH 9.0 would be sufficient to positively identify H2O2 vapours from Cl2, NO2 and O3 since only H2O2 and O3 produce a yellowing of Ti(IV) but it is only H2O2 that induces a degradation of O G at pH 9.0. The inclusion of Calmagite or indeed LG in any detector system would be useful as they generate the most rapid and visually distinctive colour changes and so provide a fast and clear indication of the presence of an oxidising vapour.
More work is needed to produce a commercially viable system, especially in making the colour change with O G and especially O II at pH 9.0 clearer and more rapid, while also attempting to completely inhibit the reactions at pH 8.0. From both a theoretical and practical point of view, it would also be useful to investigate the role of CO2 in the reactions of O G and especially O II at pH 9.0. The use of O II being a much darker shade of orange could potentially be a more useful dye to use than O G in any detection system. One attractive feature of this method is the inexpensive materials that are used and the ease of making the substrate solutions and inks. Indeed, work in these laboratories has shown that EPPS buffer, a somewhat expensive biological buffer, can be replaced by phosphate buffer for the pH 8.0 dye solutions, thus reducing the cost even further.
References
- S. Patai, The Chemistry of Peroxides, J. Wiley & Sons Ltd., New Jersey, 1983 Search PubMed.
- M. Momenteau and C. A. Reed, Chem. Rev., 1994, 94, 659 CrossRef CAS.
- F. Dubnikova, R. Kosloff, J. Almog, Y. Zeiri, R. Boese, H. Itzhaky, A. Alt and E. Keinan, J. Am. Chem. Soc., 2005, 127, 1146 CrossRef CAS PubMed.
- R. M. Burks and D. S. Hage, Anal. Bioanal. Chem., 2009, 395, 301 CrossRef CAS PubMed.
- http://www.time.com/time/nation/article/0,%208599,%201225453,00.html, accessed August 2013.
- http://www.bbc.co.uk/news/uk-12337575, accessed August 2013.
- J. Yinon, Forensic and Environmental Detection of Explosives, John Wiley & Sons Ltd., New Jersey, 1999 Search PubMed.
- H. Schubert and A. Kuznetsov, Detection of Bulk Explosives: Advanced Techniques Against Terrorism, Springer, 2004 Search PubMed.
- A. Mills, P. Grosshans and E. Snadden, Sens. Actuators, B, 2009, 136, 458 CrossRef CAS PubMed.
- H. Lin and K. S. Suslick, J. Am. Chem. Soc., 2010, 15519 CrossRef CAS PubMed.
- T. S. Sheriff, S. Cope and D. S. Varsani, Dalton Trans., 2013, 42, 5673 Search PubMed.
- L. Zhou, W. Song, Z. Chen and G. Yin, Environ. Sci. Technol., 2013, 47, 3833 CrossRef CAS PubMed.
- D. Swern, Organic Peroxides, Wiley, New York, 1970, p. 313 Search PubMed.
- S. Rothbart, E. Ember and R. van Eldik, Dalton Trans., 2010, 39, 3642 Search PubMed.
- S. I. Liochev and I. Fridovich, J. Inorg. Biochem., 2006, 100, 694 CrossRef CAS PubMed.
- A. C. Egerton, A. J. Everett, G. J. Minkoff, S. Rudrakanchana and K. C. Salooja, Anal. Chim. Acta, 1954, 10, 422 Search PubMed.
- C. E. Housecroft, and A. G. Sharpe, Inorganic Chemistry, Pearson Education Limited, 2nd edn, 2005 Search PubMed.
- P. A. Clapp, D. F. Evans and T. S. Sheriff, Anal. Chim. Acta, 1989, 218, 331 CrossRef CAS.
- http://www.h2o2.com/intro/properties/physical.html#13, accessed June 2014.
- L. Zhou, W. Song, Z. Chen and G. Yin, Environ. Sci. Technol., 2013, 47, 3833 Search PubMed.
- T. Moeller, Inorganic Chemistry: An Advanced Textbook, Wiley, New York, 1964, p. 689 Search PubMed.
Footnotes |
| † Graduated in chemistry from Queen Mary, University of London in 2013 with a 1st class honours. |
| ‡ Graduated in chemistry with forensic science from Queen Mary, University of London in 2012 with a 1st class honours. |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here. 



















