
Optical properties of xanthene based fluorescent dyes studied by stretched-film linear dichroism
Kasra Razmkhaha,
Haydn Littleb,
Sandeep Sandhuc,
Timothy R. Daffornc and
Alison Rodger*a
aDepartment of Chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: a.rodger@warwick.ac.uk; Fax: +44 (0)24 76575795; Tel: +44 (0)24 76523234
bSchool of Chemistry, University of Birmingham, Edgbaston, Birmingham, West Midlands B15 2TT, UK
cSchool of Biosciences, University of Birmingham, Edgbaston, Birmingham, West Midlands B15 2TT, UK
First published on 6th August 2014
Abstract
Xanthene dyes are commonly used to label proteins in order to probe their location and activity using fluorescence spectroscopy and microscopy. However, fundamental properties such as the polarizations of transitions for many of the dyes have not been available. In this paper we report the use of recently developed oxidised polyethylene (PEOX) stretched film linear dichroism to determine the transition polarizations of xanthene, 9-methyl-2,3,7-trihydroxy-6-fluorone, pyronin Y, pyronin B, fluorescein, and rhodamine 6G. The effect of the formation of higher order structures is also discussed when they occur. The dyes (except xanthene) all have an intense long-axis polarized transition in the region of 500 nm. They also have long-axis (//) polarized transitions from about 280 nm downwards in wavelength. There are suggestions of a weak short axis (⊥) polarized transition in the region of 320–350 nm in each case.
Introduction
It is common practice to label proteins with fluorescent dyes and use these to probe the behaviour of the proteins in microscopy and spectroscopy experiments. However, despite the extensive use of common dyes such as rhodamine 6G and fluorescein, when we came to analyse linear dichroism (LD) data for dye-labelled M13 phage we were surprised to find that fundamental properties such as the polarizations of transitions were not available.LD is the difference in absorption of light polarized parallel and perpendicular to an orientation axis.1 If the polarization of a transition in a probe molecule is known, then its LD signal can be used to determine the orientation of the probe on its host. Conversely, if the orientation is known then the transition polarization can be deduced. In this work we report the application of our new stretched film LD methods2 to a number of xanthene dyes to identify transition polarizations so that probe molecules can then be used to provide geometric information about a biomolecular assembly such as drug molecules bound to DNA3 or molecules inserted in membranes or chromophores in/on fibrous proteins4 or labels on bacteriophage.5 A complication of these and other experiments is that these planar aromatic molecules have a propensity to dimerise or form higher order oligomeric structures. So part of this work deals with that issue and its influence on the spectroscopy of the dyes.
LD may be expressed
 | (1) |
Small molecules can be oriented for LD spectroscopy in stretched polymer films.6 Polyethylene (PE) is probably the most widely used polymer film for small molecule LD as it is easy to use and has a wide spectral window. However, it cannot be used for hydrophilic molecules and PVA has traditionally been the film of choice for such molecules.1 However, PVA films take days to prepare and the analytes usually are added to the film before polymerization.7
Following work by Tissington, et al.,8 we found that oxygen plasma treatment formed oxygen containing groups on the surface of the polyethylene film thus introducing polar surface groups.2 We adopted less aggressive conditions than Tissington et al. which did not obviously affect the surface morphology. This creates a film, which we denote PEOX, that can be used even for extremely hydrophilic molecules in the same manner as PE films can be used for hydrophobic molecules. As reported in our previous paper,2 increasing the stretching factor causes an increase in the amount of the aligned crystalline phase in polyethylene leading to an enhancement in the alignment of the guest molecules deposited on the PE film. In the case of anthracene, stretching the film on which anthracene had been deposited also led into a more monomeric LD spectrum.2 In this paper we report film LD measurements on PEOX to determine the transition polarizations of some dyes that can be used as spectroscopic probes on biomolecules.
Material and methods
Materials
Xanthene and 9-methyl-2,3,7-trihydroxy-6-fluorone were obtained from Aldrich (UK) and MP Biomedicals (France) respectively, the other dyes were purchased from Sigma and used as received. Methanol and chloroform were obtained from respectively VWR Chemicals (UK) and Fisher Scientific (UK). The polyethylene film was obtained from Glad™ (Clorox Australia Pty Limited, Australia) Snap Lock plastic bags. The film was treated using an Emitech (France) K1050X Plasma Asher with parameters: O2 at 10 mmHg, 50 W RF power level, 1 min ashing time to increase the hydrophilicity of the surface.2Methods
Polyethylene films (4 cm × 2.5 cm) were cut from Glad™ Snap Lock plastic bags. PEOX pieces were then prepared by treating the films in a Emitech K1050X Plasma Asher connected to an oxygen gas supply for 1 min at 50 W power setting. They were then clamped between the jaws of a home built film2 stretcher with the initial distance between the jaws being 2.5 cm. It has been shown in general that it does not matter whether the sample is added to PE films before or after stretching,1 though different stretching amounts do affect the PE crystallite alignment and population which does affect the spectrum observed.2 However, when a dye is prone to assembling into higher order structures, different films prepared in nominally the same way can give rise to different spectra, so repeat experiments are required.The protocol used in this work to collect single LD and absorbance spectra was first to stretch the film, then add an aliquot of solvent (methanol–chloroform = 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30), measure the baseline and then add the sample and measure the sample LD and absorbance. When degree of film stretch was a variable, a baseline film and a sample film were stretched in the same way and measured for each stretch factor. Data were collected using either a Jasco (Japan) J-720 (adapted to use a 150 W Xe lamp) or a Jasco J-815 spectropolarimeter, both adapted for LD spectroscopy. The baseline spectra were subtracted from the sample spectra. Unfortunately, this methodology did not completely account for differences in scattering between the sample and baseline films, particularly in the absorbance spectra. This resulted in low wavelength LDr signals being attenuated. To calculate LDr spectra we therefore subtracted an additional baseline by zeroing spectra in linear steps at clear minima in the spectra using OriginPro 8.5.1 (OriginLab Corporation, US).
:30), measure the baseline and then add the sample and measure the sample LD and absorbance. When degree of film stretch was a variable, a baseline film and a sample film were stretched in the same way and measured for each stretch factor. Data were collected using either a Jasco (Japan) J-720 (adapted to use a 150 W Xe lamp) or a Jasco J-815 spectropolarimeter, both adapted for LD spectroscopy. The baseline spectra were subtracted from the sample spectra. Unfortunately, this methodology did not completely account for differences in scattering between the sample and baseline films, particularly in the absorbance spectra. This resulted in low wavelength LDr signals being attenuated. To calculate LDr spectra we therefore subtracted an additional baseline by zeroing spectra in linear steps at clear minima in the spectra using OriginPro 8.5.1 (OriginLab Corporation, US).
Solution absorbance spectra were obtained using a Jasco V-660 spectrophotometer with 0.1 mg mL−1 of sample dissolved in spectroscopic grade methanol. Quartz cuvettes were used with a path length of 0.01 cm. A background spectrum was measured for the solvent in the cuvette and subtracted from the sample spectrum.
Stock solutions of each dye were prepared by dissolving dye in analytical grade methanol and chloroform (70:30). An aliquot of 30 μL was dispensed onto each side of the PEOX film of desired stretch ratio (thus doubling the amount of dye while reducing the propensity to oligomerise) and the solvent was allowed to evaporate over 3 minutes in the dark under a fume hood to maintain a controlled environment and to avoid degradation of the dye.
Results and discussion
Although many of the highly fluorescing xanthene dyes often used as labels for proteins have a single aromatic ring conjugated to a xanthene motif (Fig. 1), most of their spectroscopy is due to the planar xanthene motif. We therefore began our work with the simpler structures of xanthene, 9-methyl-2,3,7-trihydroxy-fluorone (to represent the main chromophore of fluorescein), and pyronin Y and B (to represent the main chromophore of the rhodamine dyes). The key challenge in this work was the measurement of reproducible data. The dyes are prone to assembling into a range of dimer and higher order structures or aggregates. These effects were particularly a problem given the evaporation method used to produce the film sample for spectroscopy. Further, absorbance detects all species on the film, whereas LD detects only the oriented molecules making measurement of LDr challenging. To address these issues we increased the relative populations of monomers by loading less dye (though the intensities are concomitantly reduced) using more dilute stock solutions of dye. Conversely, where a population of higher order structures was required high concentration samples were loaded from higher concentration stock solutions. The degree of stretch of the film also affects the populations, with high stretch factors favouring monomers, presumably due to the increase in oriented crystallites.9 The molecules studied in this paper are all of sufficiently high symmetry that we can assume the transitions are polarized either parallel to the long axis of the xanthene motif (//) or perpendicular to it (⊥). The three-ring structures when bound to the films as isolated monomers are expected to align with their long axis along the PEOX stretch directions.1 The situations for the more complex structures (fluorescein and rhodamines) and for oligomeric structures is less obvious and are discussed below. | ||
| Fig. 1 (a) Xanthene with z and y axes indicated by // and ⊥ respectively, (b) 9-methyl-2,3,7-trihydroxy-6-fluorone, (c) pyronin Y, (d) pyronin B, (e) fluorescein, and (f) rhodamine 6G. | ||
Xanthene
Xanthene is nominally the parent compound for all the fluorescein and rhodamine dyes. Spectra for xanthene are shown in Fig. 2. There is little or no spectral shape change as a function of stretch just a gradual increase in orientation. It is clear from these data and eqn (1) that the weak transition from 280–300 nm is short axis (⊥) polarized (negative LD with LDr half the magnitude of the other bands) and the other two accessible transitions are long axis (//) polarized. | ||
| Fig. 2 Spectra of xanthene on PEOX dropped from a 0.2 mg mL−1 solution onto an unstretched film and then stretched (stretch factor as indicated in inset). // and ⊥ indicate transition polarization with respect to the xanthene long axis in Fig. 1. (a) LD, (b) LDr calculated as indicated in Methods overlaid with absorbance and LD spectra of ×2.4 stretched PEOX. | ||
9-Methyl-2,3,7-trihydroxy-6-fluorone
The fluorone chromophore of fluorescein is midway between 9-methyl-2,3,7-trihydroxy-6-fluorone (MeOH3-fluorone) and xanthene so we also analysed the spectra for MeOH3-fluorone. It is a Cs symmetry molecule as illustrated in Fig. 1, though can be treated as approximately D2h. We found it challenging to obtain a monomeric LD spectrum with reasonable signal to noise ratio for this molecule. The small LD signals also meant that scattering affected the baselines noticeably, particularly at lower wavelengths. Fig. 3a shows the overlay of two LD spectra collected on 2.4× stretched films prepared from 0.01 and 0.2 mg mL−1 stock solutions. Fig. 3b shows the overlay of corresponding absorbance and LDr spectra with a solution spectrum. The 0.01 mg mL−1 spectrum with positive LD maxima at 450 nm and 245 nm and negative maximum at 280 nm (see right hand axis of Fig. 3b) compares in position with the vibronically resolved solution phase spectrum, so we assume both are monomeric. The 450 nm and 245 nm transitions are therefore long-axis polarized (//) and 280 nm short-axis polarized (⊥). | ||
| Fig. 3 (a) LD spectra of 9-methyl-2,3,7-trihydroxy-6-fluorone on PEOX deposited from 0.01 and 0.2 mg mL−1 solution and stretched by a factor of ×2.4. // and ⊥ indicate transition polarization with respect to the xanthene long axis in Fig. 1. (b) Absorbance and LDr spectra of the LD spectra in Fig. 3(a) overlaid with solution absorbance spectrum (denoted S). | ||
The 0.2 mg mL−1 spectrum in Fig. 3a is shifted to shorter wavelength than the 0.01 mg mL−1 one with additional positive LD intensity at 400 nm. In addition a broad negative LD is apparent from 290–350 nm. We attribute the extra features to dimers or higher order assemblies of the MeOH3-fluorone. Given there is little or no extra intensity on the long wavelength side of the 450 nm band, a geometry similar to that of the so-called H-aggregate, where the monomers are stacked vertically with their long axes parallel to one another (Fig. 8a) is most likely. The 400 nm dimer signal has positive LD indicating alignment along the stretch direction, thus the dimer retains the monomer orientation. The lower wavelength regions are more complicated to interpret due largely to smaller signals and higher scattering. The 320 nm region is almost certainly short-axis polarized (⊥), whereas the 245 nm region is long axis polarized (//). It should be noted that the 0.2 mg mL−1 absorbance spectrum has evidence of structures not apparent in the LD with significant intensity above 550 nm which is presumably due to assemblies that do not orient because they are far from the polymer film.
Pyronin Y and pyronin B
Pyronin Y and B are similar C2v molecules (Fig. 1c and d) chosen for this work because of their relationship to the xanthene motif part of the rhodamine dyes. As with other dyes, the pyronins are prone to dimerization and oligomerisation and we had to use a low stock solution concentration to be able to observe the monomer spectrum.According to Jakobsen et al.,10 the pyronin Y long wavelength monomer maximum is at 547 nm with a 511 nm H-aggregate (Fig. 8a, parallel vertical stack arrangement, presumably with the substituents on alternating sides) peak.11 As shown in Fig. 4, the film LD from the most dilute pyronin Y solution has a larger positive LD component at 551 nm and a smaller one at 515 nm. When the stock solution concentration is increased and dimer or oligomers dominate the spectrum, a single positive LD band at 501 nm dominates (it is at ∼509 nm in the absorbance) with intensity at both higher and lower energy relative to the monomer. Small negative LD signals also emerge at 350 nm and 300 nm for the oligomers. These peaks may be present in the monomer spectrum, but would be below the limit of detection in the monomer spectrum due to the concentration needed to ensure monomers dominate. Dimers/oligomers also have positive LD at 254 nm. Overall we can conclude that the transitions are long axis polarized above 400 nm, short axis polarized between 300 and 400 nm, and predominantly long axis polarised below 300 nm, though of mixed polarisation below 250 nm. These conclusions are consistent with the polyvinyl alcohol film polarized absorbance and the Pariser–Parr–Pople calculations of Okubo et al.12
 | ||
| Fig. 4 (a) LD spectra of pyronin Y on PEOX deposited from solutions of concentrations indicated in the figure. // and ⊥ indicate transition polarization with respect to the xanthene long axis in Fig. 1. (b) Absorbance and LDr spectra of samples with concentrations 0.01 and 0.2 mg mL−1 overlaid with solution absorbance spectrum (S) and the dimer/monomer difference spectrum. | ||
The wavelength maximum for the pyronin B monomer is deemed to be at 552 nm and for the H-aggregates at 520 nm.11 The pyronin B LD spectra of Fig. 5 show comparatively little shape change with increasing concentration until 0.1 mg mL−1 at which point negative high-energy and positive low energy intensities become apparent. The detectable monomer absorbance signals all have positive LD so are long axis polarized. The LD thus makes it more obvious than does the absorbance that the bulky substituents of pyronin B make more complicated structure(s) than was assumed for the simple H-aggregate of pyronin Y above. Assuming that the pyronin B dimer is formed by associating a second monomer with one already aligned on the film, a geometry giving rise to an in-phase positive exciton component at longer wavelength and an out-of-phase negative component at shorter wavelength2 is to stack the pyronins as in a brick wall thus somewhere between an H- and J-aggregate as illustrated in Fig. 8b. This geometry makes room for the bulky substituents and allows for effective π–π interactions.
 | ||
| Fig. 5 (a) LD spectra of pyronin B on PEOX deposited from solutions of concentrations indicated in the figure. // and ⊥ indicate transition polarization with respect to the xanthene long axis in Fig. 1. (b) Absorbance and LDr spectra of samples with concentrations 0.01 and 0.2 mg mL−1 overlaid with solution absorbance spectrum (S) and the dimer/monomer difference spectrum. | ||
Fluorescein
Film LD spectra for fluorescein are given in Fig. 6. The low concentration sample (0.01 mg mL−1) has positive LD at 497 nm and 465 nm, perhaps a very small negative LD at 322 nm, positive LD at 277 nm, and positive LD at 230 nm in accord with literature positions for monomer absorbances.13,14 Thus, as with fluorone, we conclude that: the longest wavelength band is polarized along the long axis (//) of the xanthene chromophore; there is probably a short axis polarized (⊥) transition at 322 nm; and the next bands are long axis polarized. At even slightly higher concentrations, the film LD spectrum becomes much more complicated with evidence of a small negative LD at 560 nm, positive LD at 516 nm, and negative LD at 453 nm with probably another component between these two. The lower wavelength region also changes. The number of new bands suggests a variety of oligomeric structures are formed on the film. The fluorescein oligomeric spectra resemble those of pyronin B suggesting that fluorescein can also stack on the PE film in the brick-like structure of Fig. 8b.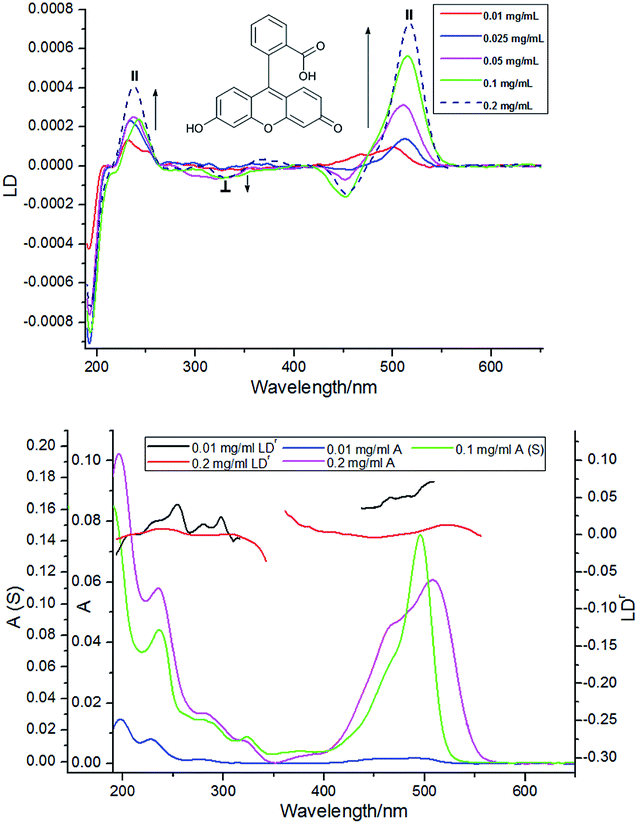 | ||
| Fig. 6 (a) LD spectra of fluorescein on PEOX deposited from solutions of concentrations indicated in the figure. // and ⊥ indicate transition polarization with respect to the xanthene long axis in Fig. 1. (b) Absorbance and LDr spectra of samples with concentrations 0.01 and 0.2 mg mL−1 overlaid with solution absorbance spectrum (S) and the dimer/monomer difference spectrum. | ||
Rhodamine 6G
Fig. 7 shows the film LD and absorbance spectra of rhodamine 6G as a function of concentration of stock solution. At low concentration there is a larger positive LD peak at 535 nm and smaller positive peaks at 350, 290, 248, and 229 nm indicating these are all polarized along the long axis (//) of the xanthene chromophores. Comparison of solution absorbance data15 with the film spectra indicates that this spectrum is monomeric rhodamine 6G. The high concentration samples behave similarly to the fluorescein, though have a greater propensity to stack. | ||
| Fig. 7 (a) LD spectra of rhodamine 6G on PEOX deposited from solutions of concentrations indicated in the figure. // and ⊥ indicate transition polarization with respect to the xanthene long axis in Fig. 1. (b) Absorbance and LDr spectra of samples with concentrations 0.01 and 0.2 mg mL−1 overlaid with solution absorbance spectrum (S) and the dimer/monomer difference spectrum. | ||
Conclusions
We have undertaken careful studies of xanthene dyes on oxidised polyethylene film in order to be able to assign the polarizations of the component transitions. By depositing samples from 0.01 mg mL−1 solutions we were able to measure monomer stretched film spectra. Although xanthene is nominally the parent compound of these dyes, its spectroscopy differs significantly from the others. It has a small negative (short axis polarized, ⊥) LD band at ∼290 nm and its higher energy transitions are long axis (//) polarised. The extra conjugation introduced into the xanthene moiety of fluorones, pyronins, fluoresceins and rhodamines results in an intense long-axis polarized transition in the region of 500 nm. They also have long-axis (//) polarized transitions from about 280 nm downwards in wavelength. There are suggestions of a weak short axis (⊥) polarized transition in the region of 320–350 nm in each case.Upon depositing the dyes from high concentration solutions, with the exception of xanthene itself, the spectra changed shape showing intensity on either the high energy side or on both high and low energy sides of the lowest energy transition indicating exciton coupling of the dyes. 9-Methyl-2,3,7-trihydroxy-6-flourone and pyronin Y spectra are suggestive of them forming H-aggregates (parallel vertical stacks, Fig. 8a) aligned along the film stretch direction. This is consistent with the literature for pyronin Y. Pyronin B has also been suggested to form H-aggregates in solution, however, on the films we observed both high and low energy exciton components whose high energy term was negative in sign. A geometry that gives this sign pattern is an offset H-aggregate like a portion of a brick wall aligned along the film stretch direction (Fig. 8b). We postulate this is the geometry adopted in the pyronin B dimers/oligomers. Fluorescein and rhodamine 6G both follow the pyronin B spectroscopy so may also be adopting a brick-like stacking, presumably driven by the need to avoid the extra substituents overlapping.
 | ||
| Fig. 8 (a) H-aggregate geometry which is consistent with the data for MeOH3-fluorone and pyronin Y at high concentrations. (b) Offset stacked geometry which is consistent with the data for pyronin B, fluorescein, and rhodamine 6G at high concentrations. | ||
References
- B. Nordén, A. Rodger and T. R. Dafforn, Linear dichroism and circular dichroism: a textbook on polarized spectroscopy, Royal Society of Chemistry, Cambridge, 2010 Search PubMed.
- K. Razmkhah, M. I. Gibson, N. P. Chmel and A. Rodger, Analyst, 2014, 139, 1372 RSC.
- C. Uerpmann, J. Malina, M. Pascu, G. J. Clarkson, V. Moreno, A. Rodger, A. Grandas and M. J. Hannon, Chem.–Eur. J., 2005, 11, 1750 CrossRef CAS PubMed.
- E. Small, R. Marrington, A. Rodger, D. J. Scott, K. Sloan, D. Roper, T. R. Dafforn and S. G. Addinall, J. Mol. Biol., 2007, 369, 211 CrossRef PubMed.
- R. Pacheco-Gomez, J. Kraemer, S. Stokoe, H. J. England, C. W. Penn, E. Stanley, A. Rodger, J. Ward, M. R. Hicks and T. R. Dafforn, Anal. Chem., 2012, 84, 91 CrossRef CAS PubMed.
- E. W. Thulstrup, J. Michl and J. H. Eggers, J. Phys. Chem., 1970, 74, 3868 CrossRef CAS.
- M. A. Ismail, K. J. Sanders, G. C. Fennel, H. C. Latham, P. Wormell and A. Rodger, Biopolymers, 1998, 46, 127 CrossRef CAS.
- B. Tissington, G. Pollard and I. M. Ward, Compos. Sci. Technol., 1992, 44, 185 CrossRef CAS.
- A. Wirtz, C. Hofmann and E. Groenen, ChemPhysChem, 2011, 12, 1519 CrossRef CAS PubMed.
- P. Jakobsen, H. Lyon and S. Treppendahl, Histochemistry, 1984, 81, 99 CAS.
- M. Arık, K. Meral and Y. Onganer, J. Lumin., 2009, 129, 599 CrossRef PubMed.
- J. Okubo, I. Ono, T. Hoshi, A. Yoshitake, M. Kobayashi and N. Inoue, J. Soc. Photogr. Sci. Technol. Jpn., 1991, 54, 150 CAS.
- I. L. Arbeloa, J. Chem. Soc., Faraday Trans. 2, 1981, 77, 1725 RSC.
- R. Sjoback, J. Nygren and M. Kubista, Spectrochim. Acta, Part A, 1995, 51, L7 CrossRef.
- (a) Y. Lu and A. Penzkofer, Chem. Phys., 1986, 107, 175 CrossRef CAS; (b) J. E. Selwyn and J. I. Steinfeld, J. Phys. Chem., 1972, 76, 762 CrossRef CAS; (c) P. Bojarski, A. Matczuk, C. Bojarski, A. Kawski, B. Kuklinski, G. Zurkowska and H. Diehl, Chem. Phys., 1996, 210, 485 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |