
Facile synthesis of few-layer-thick carbon nitride nanosheets by liquid ammonia-assisted lithiation method and their photocatalytic redox properties†
Ying Yina,
Jiecai Hana,
Xinghong Zhanga,
Yumin Zhanga,
Jigang Zhouc,
David Muirc,
Ronny Sutartoc,
Zhihua Zhangd,
Shengwei Liu*e and
Bo Song*ab
aCenter for Composite Materials, Harbin Institute of Technology, Harbin 150080, China. E-mail: songbo@hit.edu.cn
bAcademy of Fundamental and Interdisciplinary Sciences, Harbin Institute of Technology, Harbin 150080, China
cCanadian Light Source Inc., Saskatoon, Saskatchewan S7N 0X4, Canada
dLiaoning Key Materials Laboratory for Railway, School of Materials Science and Engineering, Dalian Jiaotong University, Dalian 116028, China
eState Key Laboratory of Advanced Technology for Materials Synthesis and Processing, Wuhan University of Technology, Wuhan 430070, China. E-mail: sliu@whut.edu.cn
First published on 21st July 2014
Abstract
High-quality few-layer-thick graphitic carbon nitride (g-C3N4) nanosheets (NSs) were fabricated by a simple, highly efficient, and rapid method namely, liquid ammonia (LA)-assisted lithiation. Li intercalation occurred in less than half an hour, importantly, the degree of Li intercalation was indicated by the color change of LA solution from deep blue to colorless. The obtained products were carefully investigated by field-emission transmission electron microscopy, field-emission scanning electron microscopy, atomic force microscopy, X-ray powder diffraction, X-ray photoelectron spectroscopy, Raman scattering spectrometry, UV-visible absorption spectrometry, photoluminescence, soft X-ray absorption and nonresonant soft X-ray emission spectroscopy, and X-ray absorption near-edge structure analyses. Because of the lack of high-temperature or high-energy treatment, high-yield few-layer-thick g-C3N4 NSs were produced with trace O2 impurity. Interestingly, while maintaining the similar crystal structure and chemical stoichiometric ratio relative to the parent bulk materials, the surface structure, electronic and optical properties were significantly varied. Moreover, compared to the bulk counterparts, the as-prepared g-C3N4 NSs show clearly enhanced photocatalytic redox activity with respect to both photocatalytic H2 evolution and hydroxyl radical generation. LA-assisted lithiation is a general method and could be easily extended to exfoliate diverse other layered materials such as molybdenum and tungsten sulfides.
1. Introduction
Two-dimensional (2D) NSs have attracted tremendous attention owing to their unusual properties resulting from their unique surface atomic geometry and electronic structure.1–3 As a typical example, graphene, a 2D single layer of carbon atoms, has demonstrated exceptional properties and a wide range of applications in nanoelectronics, photodetectors, capacitors, and catalysis.4–6 These have triggered keen interests to synthesize atomic-thick NSs other than graphene and investigate their properties. To date, inspired by the available routes to fabricate graphene and graphene oxide, several ultrathin 2D NSs have been obtained by diverse methods such as mechanical cleavage,7 ball milling,8 ultrasonication in common solvents,9 and ion intercalation.10,11 Unfortunately, these available pathways are limited in regards to yield, purity and efficient. For instance, Ramakrishna et al. use the n-butyl lithium in hexane as the intercalation agent to insert lithium ions and then ultrasonication in water to peel off the nanosheet3 Nevertheless, long reaction time (e.g. 3 days) and poor controllability over lithium insertion process restrict its popularization. Alternatively, electrochemical lithiation has been used to exfoliate MoS2, WS2, etc.10 which is inconvenient to manipulate (complicated fabrication of lithium battery) and high output is inaccessible. Despite the scientific and technological importance and significant efforts, the progress in this field is still limited. Therefore, a facile, effective, and versatile method for the fabrication of single- or few-layer ultrathin 2D NSs is highly desirable and is of special interest owing to its unique and fascinating properties, providing additional opportunities for the applications of 2D-layered materials.Recently, as a new type of 2D-layered material, metal-free graphitic carbon nitride (g-C3N4) NSs have received increasing attention.12–14 In particular, this fascinating semiconductor with a bandgap of ∼2.64 eV has been intensively investigated as an alternative metal-free visible-light-responsive photocatalyst.15–21 g-C3N4 has a graphite-like structure with strong C–N covalent bonding in the in-plane direction and weak van der Waals forces between the C–N layers with a layer distance of ∼3.3 nm.15–21 This intrinsically layered structure allows to exfoliate bulk materials to afford a monolayer or atomically thick g-C3N4 NS.12–14 Compared to the bulk g-C3N4, the highly anisotropic 2D NSs may possess a much higher specific surface area, a larger bandgap because of the quantum size effect, improved electron transport ability along the in-plane direction, and increased lifetime of photoexcited charge carriers because of a higher separation efficiency.12–14 Liu et al. reported that g-C3N4 NSs with a thickness of ∼2 nm can be obtained by the thermal oxidation etching of bulk g-C3N4 in air.12 However, the thermal oxidation etching afforded only ∼6 wt% yield.9 Zhang et al. reported a water-mediated exfoliation method to prepare g-C3N4 NSs.13 The exfoliation process was significantly influenced by the surface energy of the solvent molecules. Yang et al. reported an isopropanol-mediated exfoliation method to obtain g-C3N4 NSs with enhanced performance in photocatalytic H2 evolution.14 Although solvent-mediated exfoliation methods are feasible, their efficiency is low. Therefore, it is still challenge to develop an effective method to fabricate few-layer-thick g-C3N4 NSs.
Herein, we report a simple and efficient method to fabricate few-layer-thick g-C3N4 NSs by a LA-assisted Li intercalation method. Because of the high surface/bulk ratio, the as-prepared g-C3N4 NSs exhibited enhanced photocatalytic performance compared to their bulk counterpart. Significantly, this LA-assisted lithiation method is versatile and effective to exfoliate other 2D-layered materials such as MoS2 and WS2 in a large scale and high yield, providing additional opportunities to meet the intense demand for practical catalytic, biological, and electrical applications. To the best of our knowledge, this is the first report on the synthesis of few-layer-thick 2D g-C3N4, MoS2, and WS2 NSs by the LA-assisted lithiation method.
2. Experimental
2.1 Preparation of bulk g-C3N4
Melamine (10 g, Alfa Aesar, 99.999%) was heated at 550 °C for 2 h under the protection of 20 standard cubic centimeter per minute (sccm) N2 (99.999%) with a ramp rate of 3 °C min−1 for both heating and cooling processes, affording yellow g-C3N4 powder (yield: ∼3 g).2.2 Preparation of few-layer-thick g-C3N4 NSs
First, the Li intercalation of g-C3N4 was carried out in LA using a Schlenk line (see ESI Fig. S1†). 0.3 g as-prepared bulk g-C3N4 yellow powder and 0.01 g of Li pieces (Alfa Aesar, 99.99%) (molar ratio, 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) were placed at the bottom of a silica tube. This process was performed in an argon-filled glove box (Mbraun, Unilab, Germany) to prevent air and water contamination. Then, the silica tube was placed in a bath of 70:30 alcohol–ice water mixture, which could stabilize the temperature at −48 °C, and evacuated to a vacuum of 10−2 Pa. High purity ammonia (99.9999%) gas was introduced into the tube and condensed into liquid (up to 12 mL) in which the yellow g-C3N4 powder was immersed. Then, the reaction was by shaking the tube, thus contacting the LA with Li; the blue color (characteristic color of e−(NH3)n) gradually faded within 30 min. Finally, the ammonia was carefully removed by evaporation. After the intercalation, the Li-intercalated sample was exfoliated and ultrasonicated in deionized (DI) water for 30 min. During this process, a large number of bubbles were observed, and an opaque suspension of the product was obtained. After the suspension was centrifuged at 3000 rpm to remove the residual unexfoliated g-C3N4 particles and washed five times with DI water, the products was characterized. The preparation of MoS2 and WS2 NSs is similar to that of g-C3N4 NSs as mentioned above.
:1) were placed at the bottom of a silica tube. This process was performed in an argon-filled glove box (Mbraun, Unilab, Germany) to prevent air and water contamination. Then, the silica tube was placed in a bath of 70:30 alcohol–ice water mixture, which could stabilize the temperature at −48 °C, and evacuated to a vacuum of 10−2 Pa. High purity ammonia (99.9999%) gas was introduced into the tube and condensed into liquid (up to 12 mL) in which the yellow g-C3N4 powder was immersed. Then, the reaction was by shaking the tube, thus contacting the LA with Li; the blue color (characteristic color of e−(NH3)n) gradually faded within 30 min. Finally, the ammonia was carefully removed by evaporation. After the intercalation, the Li-intercalated sample was exfoliated and ultrasonicated in deionized (DI) water for 30 min. During this process, a large number of bubbles were observed, and an opaque suspension of the product was obtained. After the suspension was centrifuged at 3000 rpm to remove the residual unexfoliated g-C3N4 particles and washed five times with DI water, the products was characterized. The preparation of MoS2 and WS2 NSs is similar to that of g-C3N4 NSs as mentioned above.
2.3 Characterization
X-ray diffraction (XRD) data were collected using a high-resolution powder diffractometer (Rigaku D/max 2500, CuKα, λ = 0.15418 nm) at room temperature. Raman scattering measurement was performed at room temperature using a Raman system (JY-HR800) equipped with a 532 nm line from a solid-state laser. An FEI Sirion-200 field-emission scanning electron microscopy (SEM) and a JEM-2100F field-emission transmission electron microscopy (TEM) operating at 200 keV were used to characterize the synthesized materials. The point resolution of the high-resolution TEM (HRTEM) was ∼0.19 nm. The thickness of the material was analyzed by atomic force microscopy (AFM) using a Bruker DI MultiMode-8 system. For the chemical compositions analysis, the samples were analyzed using a K-alpha X-ray photoelectron spectroscopy (XPS) system using an Al Kα X-ray source and the binding energy values were measured with respect to the C 1s peak at 283.5 eV. For the optical measurement, UV-visible absorption spectra were recorded using a UV-visible spectrophotometer (UV2550, Shimadzu, Japan), and photoluminescence (PL) spectroscopy was measured using a Horiba JY HR800 instrument. N2 adsorption–desorption isotherms were measured using an F-sorb X400 surface area analyzer. All the samples were outgassed at 150 °C for 10 h prior to the N2 adsorption measurements. Soft X-ray absorption (XAS) and nonresonant soft X-ray emission spectroscopy (XES) spectroscopic measurements of C and N K-edges were conducted using the new XES endstation that is currently being commissioned on the resonant elastic and inelastic X-ray scattering (REIXS) beamline at the Canadian Light Source (CLS). The Rowland circle spectrometer with a microchannel plate imaging detector on that endstation was configured for an approximate resolving power of 103 for the XES measurements. The incident light was supplied by the monochromator with a resolution of better than 5 × 103 for the XAS measurements, and the data were collected using total electron yield (TEY). The XAS and XES measurements were calibrated by graphite (285.4 eV) and h-BN (400.9 eV) at C and N edges, respectively. The X-ray absorption near-edge structure (XANES) measurements at C K and N K-edges were collected at the spherical grating monochromator (SGM) beamline at the CLS, the University of Saskatchewan, Canada.2.4 Photocatalytic redox properties
The photocatalytic H2 production experiments were performed in a 100 mL 3-neck Pyrex flask at ambient temperature and atmospheric pressure, and the outlets of the flask were sealed with silicone rubber septums. A 350 W xenon arc lamp, which was positioned 20 cm away from the reactor, was used as the light source to trigger the photocatalytic reaction. The focused intensity on the flask was ∼20 mW cm−2. In a typical photocatalytic experiment, 50 mg of the sample was dispersed in 80 mL of an aqueous solution containing 10% v/v triethanolamine scavenger. The decomposition of 6 wt% Pt catalyst was conducted by directly dissolving H2PtCl6 into the above 300 mL solution. Before the irradiation, the system was bubbled with N2 for 40 min to remove the dissolved O2, ensuring anaerobic condition. H2 was analyzed using a gas chromatograph (GC-14C, Shimadzu, Japan, TCD, N2 as the carrier gas and 5 Å molecular sieve column). Hydroxyl radical (˙OH) reactions were performed as follows: 5 mg sample was suspended in 80 mL of an aqueous solution containing 0.01 M NaOH and 3 mM terephthalic acid. Before the exposure to irradiation, the suspension was stirred in dark for 30 min. Then, 5 mL of the solution was taken out every 10 min under λ > 400 nm and centrifuged for fluorescence spectrum measurements. During the photoreactions, no O2 was bubbled into the suspension. The fluorescence signal of the generated 2-hydroxy terephthalic acid was measured using a Hitachi F-4500 fluorescence spectrophotometer with an excitation wavelength of 320 nm.3. Results and discussion
The real-time Li intercalation of bulk g-C3N4 in LA is shown in Fig. 1. Initially, bulk g-C3N4 powder exhibits yellow color (Fig. 1a), while the LA solution with dissolved Li in silica tube exhibits deep indigo blue color (Fig. 1b). After mixing them and shaking for 10 min, the Li ions gradually inserted into the g-C3N4, and the color of the resulting LA solution gradually faded to deep green (Fig. 1c). The color of the solution continually faded during the shaking, became colorless after 30 min (Fig. 1d, the yellow color originates from the suspended g-C3N4 fragments), indicating that most of the Li ions intercalated into the g-C3N4 NSs and small amounts of Li ions are left in the LA solution. Next, the lithium-intercalated g-C3N4 was ultrasonicated in DI water prior to its exfoliation into a well-dispersed transparent NS suspension. | ||
| Fig. 1 Real-time Li intercalation of bulk g-C3N4 in LA. | ||
Fig. 2a shows the TEM image of the as-prepared samples; which are transparent to electron beams because of their ultrathin thickness, indicating that NSs were obtained. The inset in Fig. 2a shows the HRTEM image of the as-obtained NSs. The obtained lattice spacing of 0.35 nm was assigned to (100) plane, confirming that the products still contain hexagonal g-C3N4 (ICDD-PDF-4+ no. 00-04-0836) as the bulk counterparts. The energy-dispersive X-ray spectroscopy (EDS), with a detection limit of 1–2 at. %, of the as-prepared g-C3N4 NSs (Fig. 2b) detected mainly C and N elements with a nominal atomic ratio of 3:4 without the presence of Li in the samples, indicating that metal Li metal only helps to exfoliate the layered structures and eliminated completely. The observed trace O element may have originated from the absorption of O2 or H2O when the as-prepared samples were exposed to air. The thickness of the as-prepared g-C3N4 NSs was measured by AFM (Fig. 2c); clearly, the height of a random NS fragment is ∼2.5 nm (Fig. 2d), which is approximately seven C–N layers in contrast to its parent (bulk) source consisting of hundreds of layers, indicating that few-layer-thick g-C3N4 NSs were obtained. Notably, the as-prepared few-layer-thick g-C3N4 NSs well dispersed in DI water even after storing for one month under ambient conditions (ESI, Fig. S2†). Further, the chemical composition of g-C3N4 NS was investigated by energy-filtered TEM (Fig. 3 and ESI, Fig. S3†). The uniform distribution of C and N elements confirms that this method facilitates a nondestructive exfoliation of g-C3N4, and no residual Li element was detected. Further, the SEM images (ESI, Fig. S4†) show that the as-exfoliated g-C3N4 NSs maintained the same lateral scale range as the parent (bulk) g-C3N4, but show a few-layer-thick feature in thickness.
 | ||
| Fig. 2 (a) TEM image of g-C3N4 NS. Inset shows the HRTEM image of the selected area indicated by a box. (b) EDS of the as-exfoliated g-C3N4 NSs deposited on a Cu foil. (c) AFM image of the as-exfoliated g-C3N4 NSs. (d) The corresponding height image of a random g-C3N4 NS. | ||
 | ||
| Fig. 3 Energy-filtered TEM images of C and N elements in the as-prepared g-C3N4 NSs. | ||
To better understand the crystal structure, the as-prepared g-C3N4 NSs were characterized by XRD as shown in Fig. 4. Clearly, the exfoliation of few-layer-thick g-C3N4 NSs from bulk g-C3N4 induced considerable change in the XRD pattern. In contrast to bulk g-C3N4, the typical diffraction peak at 13.1°, which stems from the lattice planes parallel to the c-axis, disappeared in the XRD pattern of g-C3N4 NSs. Moreover, the intensity of the diffraction peak at 27.6° corresponding to the (002) plane decreased remarkably. Both these changes confirm that the bulk g-C3N4 was successfully exfoliated into few-layer-thick NSs, consistent with the abovementioned characterization results.
 | ||
| Fig. 4 XRD patterns of bulk g-C3N4 and g-C3N4 NSs. Inset shows a schematic diagram of the fabrication of the g-C3N4 NSs by LA-assisted lithiation. | ||
The electronic structure of the as-prepared g-C3N4 NSs was investigated by the combined analysis of optical absorption and PL spectra. As shown in Fig. 5a, the intrinsic absorption peak of g-C3N4 NSs shows a distinct blue shift of ∼10 nm compared to bulk g-C3N4.12 The PL spectra show a clear blue shift from 459 nm (bulk g-C3N4) to 449 nm (g-C3N4 NSs) (Fig. 5b). Both these typical blue shifts can be attributed to the quantum confinement effect resulting from the shifting of conduction and valence bands in opposite directions.13,14 In addition, g-C3N4 NSs shows a stable PL peak without apparent shift under different excitation wavelength (Fig. S8†) and varied excitation intensity (Fig. S9†), implying its intrinsic stimulated emission mechanism.
 | ||
| Fig. 5 (a) UV-visible absorption and (b) fluorescence emission spectra of g-C3N4 NS and bulk g-C3N4. | ||
It is well established that XANES is highly sensitive and can be used to determine the changes in the valence of atoms in a matrix. To further investigate the electronic and chemical structures of the as-prepared g-C3N4 NSs, C and N K-edge XANES and XES spectra of the g-C3N4 NSs were obtained using the bulk g-C3N4 as the reference. The identical XES and similar XANES spectra (Fig. 6a) between the g-C3N4 NSs and bulk matrix proved that the as-prepared g-C3N4 NSs maintained the major structure as the bulk material. The XES spectra of g-C3N4 (Fig. 6a), both NSs and bulk material, are close to that of graphite, but apparently different from crystalline β-C3N4 because of the difference in bonding.16
 | ||
| Fig. 6 (a) XES and XANES C K-edge for g-C3N4 NSs and bulk g-C3N4. (b) XAS N K-edge for g-C3N4 NSs and bulk g-C3N4. (c) XES measurements of g-C3N4 NSs and bulk g-C3N4 at the N K-edge. | ||
The XANES spectrum of g-C3N4 at the C K-edge is composed of a sharp π* (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) transition at ∼288 eV and a broad σ* peak at ∼294 eV. The N K-edge XANES spectra (Fig. 6b) are composed of a broad σ* transition peak located at 407 eV and two π* peaks located at 399 and 402 eV, which have been assigned to the pyridine- and graphite-like N bonding in g-C3N4, respectively.22,23 Interestingly, the surface-sensitive TEY mode XANES measurements show clear structural differences in the g-C3N4 NSs than bulk-sensitive XES measurements. The g-C3N4 NSs exhibit a stronger π* feature in both the C and N K-edge XANES spectra, predicting a better electronic conductivity. Furthermore, the σ* peak in the C K-edge XANES in the g-C3N4 NSs shifted to a higher energy direction, indicating that the shortening of C–N bonds in plane was probably induced by the presence of a strain in g-C3N4 NSs after the exfoliation, and similar results have been observed in graphene.24,25 Moreover, the XANES spectra of g-C3N4 NSs at the C and N K-edges show a lower-energy shoulder beside the first π* orbital, indicating the presence of more dangling bonds in g-C3N4 NSs. The spectrum of g-C3N4 NSs is slightly broader with higher intensity, indicating that the g-C3N4 NSs have better electronic conductivity (Fig. 6c). Notably, the band structure determined by XES and XAS is seemingly different from the results of UV-visible and PL measurements. The UV-visible result (Fig. 5a) indicates that the bandgap widens, while the XES and XAS results indicate that the bandgap narrows. A possible reason for this discrepancy is that XAS at TEY is surface sensitive (<5 nm deep), while XES is bulk sensitive. All these results confirm a rich electronic structure modification in g-C3N4 NSs but mostly confined to surface, while the g-C3N4 NS sample still retained the same structure as the bulk counterpart.
N) transition at ∼288 eV and a broad σ* peak at ∼294 eV. The N K-edge XANES spectra (Fig. 6b) are composed of a broad σ* transition peak located at 407 eV and two π* peaks located at 399 and 402 eV, which have been assigned to the pyridine- and graphite-like N bonding in g-C3N4, respectively.22,23 Interestingly, the surface-sensitive TEY mode XANES measurements show clear structural differences in the g-C3N4 NSs than bulk-sensitive XES measurements. The g-C3N4 NSs exhibit a stronger π* feature in both the C and N K-edge XANES spectra, predicting a better electronic conductivity. Furthermore, the σ* peak in the C K-edge XANES in the g-C3N4 NSs shifted to a higher energy direction, indicating that the shortening of C–N bonds in plane was probably induced by the presence of a strain in g-C3N4 NSs after the exfoliation, and similar results have been observed in graphene.24,25 Moreover, the XANES spectra of g-C3N4 NSs at the C and N K-edges show a lower-energy shoulder beside the first π* orbital, indicating the presence of more dangling bonds in g-C3N4 NSs. The spectrum of g-C3N4 NSs is slightly broader with higher intensity, indicating that the g-C3N4 NSs have better electronic conductivity (Fig. 6c). Notably, the band structure determined by XES and XAS is seemingly different from the results of UV-visible and PL measurements. The UV-visible result (Fig. 5a) indicates that the bandgap widens, while the XES and XAS results indicate that the bandgap narrows. A possible reason for this discrepancy is that XAS at TEY is surface sensitive (<5 nm deep), while XES is bulk sensitive. All these results confirm a rich electronic structure modification in g-C3N4 NSs but mostly confined to surface, while the g-C3N4 NS sample still retained the same structure as the bulk counterpart.
The composition and chemical states of the g-C3N4 NSs were also investigated by XPS (ESI, Fig. S5†). The C 1s peak located at ∼283.5 eV originates from the standard reference C, while that located at ∼287.5 eV represents the sp2-bonded C of g-C3N4.15–21 The XPS results indicate that the as-exfoliated g-C3N4 NSs are of high purity and mainly composed of C and N. Compared to the bulk g-C3N4, the binding energy of the g-C3N4 NSs shifted to a higher binding energy because of the size effect. The O content slightly increases from 3% (bulk g-C3N4) to 5% (g-C3N4 NSs), and the tiny amount of oxygen element in bulk g-C3N4 and g-C3N4 NSs could be ascribed to the tiny amount of O2 or H2O adsorbed on the surface when exposed to the air or during the ultrasonication process in deionized water, which is a common phenomenon in the synthetic g-C3N4.13 The atomic ratio of C to N decreased from 0.74 in the bulk g-C3N4 to 0.69 in the g-C3N4 NSs (ESI, Table S1†), consistent with that reported by Zhang et al. using an ultrasonication exfoliation method.13 This indicates that high-quality g-C3N4 NSs were obtained by the LA-assisted lithiation method. To further investigate the structural feature of the as-exfoliated g-C3N4 NSs, Raman measurement was performed (ESI, Fig. S6†). The g-C3N4 NSs exhibited almost the same Raman modes as their bulk counterpart, indicating that the exfoliated ultrathin g-C3N4 NSs retained the same crystal structure as the bulk counterpart. Notably, no clear blue shift was observed in the Raman spectra of g-C3N4 NSs,13 probably because of the identical lateral scale as the bulk g-C3N4 (ESI, Fig. S4†) in which the phonon confinement effect does not act differently. Moreover, for both the g-C3N4 bulk and NS materials, there are no shifts in the C peak, which was located at ∼1350 and 1580 cm−1, respectively, confirming the high purity of the as-exfoliated g-C3N4 NSs.
Significantly, this LA-assisted lithiation method can also be applied to exfoliate diverse 2D-layered material such as MoS2 and WS2. The AFM analyses (ESI, Fig. S7†) of the as-prepared MoS2 and WS2 NSs showed an average thickness of ∼3.5 and ∼3.0 nm, respectively, confirming that few-layer-thick MoS2 and WS2 NSs were successfully obtained. It is well known that LA is transparent and colorless in nature, while Li metal is dissolved in LA to generate metal cations, Li+ and solvated electron, e−(NH3)n, exhibiting a deep blue color arising from the presence of solvated electrons.26–34 Therefore, the entire process of Li insertion into the g-C3N4 matrix can be visualized by the color fading of the LA solution. The basic reaction corresponding to Fig. 1b can be described by eqn (1),
| Li + NH3(l) → Li+ + e−(NH3)n | (1) |
It is proposed that, in addition to Li+ ion diffusion, the Li+ ions probably penetrated the C3N4 interlayers (Step 1, Fig. 7), involving the in situ redox reactions catalyzed by LA, which can be described by eqn (2).
| xLi+ + C3N4(bulk) → xLi + C3N4+ | (2) |
 | ||
| Fig. 7 Schematic diagram of the lithiation and exfoliation of g-C3N4 NSs from bulk g-C3N4. | ||
It is rationalized that the lithiation was much faster than the traditional Li intercalation only based on the diffusion of Li+ ions.3 These Li+ located between the interlayers to form a charged g-C3N4 material, which was subsequently compensated by the solvated electrons to form a neutral g-C3N4 material with intercalated metallic Li during the shaking, as shown by eqn (3)
| xLi + C3N4+ + e−(NH3)n → Li@C3N4(bulk) | (3) |
During these processes, the characteristic deep blue color of e−(NH3)n gradually faded because of the decrease in e−(NH3)n concentration, corresponding to Fig. 1c. After the insertion of metallic Li into the interlayers, the interlayer distance increased, thus weakening the van der Waals interactions between the layers (Step 2, Fig. 7). To examine the possible reaction between g-C3N4 and ammonia, the structure of bulk g-C3N4 before and after soaked in ammonia (without lithium) was compared by XRD analysis (ESI, Fig. S11†). It can be seen that no crystal structure change was found, implying bulk g-C3N4 is inert to liquid ammonia. Next, the products were exfoliated in DI water by ultrasonication (Step 3, Fig. 7). In this step, metallic Li reacts with H2O to form LiOH and H2, thus pushing the layers apart from each other. In fact, a large number of bubbles were observed during the exfoliation. Thus, isolated few-layer-thick g-C3N4 NSs were obtained. The corresponding reaction process can be described by eqn (4).
| Li@C3N4 + H2O → C3N4(NS) + LiOH + H2↑ | (4) |
Notably, the as-proposed LA-assisted lithiation method is better than those reported by Zhang et al. and Kaner et al.10,26,27 owing to five outstanding merits of our study: (1) in particular, the large-scale production (∼10 g) of g-C3N4, MoS2, and WS2 NSs (for one time) has been achieved with a high yield of ∼85%. (2) Compared to the conventional methods, this method is less time-consuming. Most of the reactions were completed in <30 min; for MoS2, this process occurred in ∼5–10 min. (3) A significant feature of this method is that the degree of Li intercalation can be visually observed from the clear color changes in the LA solution from deep blue color to colorless, while the Li intercalation process can be tuned by the shaking speed of the silica tube. (4) In the absence of high-temperature treatment or oxidation process, the as-prepared NSs obtained by a low-temperature synthesis exhibited the same crystal structure and chemical stoichiometric ratio of parent (bulk) materials, exhibiting the intrinsic features of the parent (bulk) material. (5) The LA-assisted lithiation method also expands the range of layered materials that can be inserted by metallic Li than the special one.34–36 Moreover, it should be noted that the metals, Na and K, can also be used to exfoliate the 2D-layered compounds by the similar route. Thus, this route can be defined as LA-assisted alkali metal intercalation strategy.
The intriguing surface characteristics, electronic and electrical properties, and optical responses of the as-prepared g-C3N4 NSs make it promising for diverse applications. As a proof of concept, the advantage of g-C3N4 NSs as a photocatalyst was demonstrated in this study. The photocatalytic activity of the as-prepared g-C3N4 NSs was first evaluated in the photocatalytic H2 evolution from a water/triethanolamine solution. Under visible light irradiation, while a trace amount of H2 was detected using bulk g-C3N4 as the photocatalyst, the as-prepared g-C3N4 NSs showed extensively enhanced photocatalytic H2 evolution at a steady rate (Fig. 8a), indicating the stability of g-C3N4 NSs as photocatalysts. Here, the as-prepared g-C3N4 NS exhibited an average hydrogen evolution rate of ∼8 μmol h−1 within 0–5 h, which is about twice than that of bulk parent material as obtained in Wang et al.' work.15 Moreover, the g-C3N4 NSs also showed great ability in activating H2O molecules to produce highly reactive hydroxyl radicals (Fig. 8b), which can be used as reactive oxidative species to trigger various oxidation reactions for environmental remediation and organic transformations. The enhanced photocatalytic activity of g-C3N4NS can be attributed to the increased surface area (ESI, Fig. S10†), abundant dangling bonds, and modified electronic structures as demonstrated previously. The high surface/bulk ratio and more dangling bonds in g-C3N4 NSs provide abundant reactive sites. The few-layer-thick thickness and better electronic conductivity shorten the bulk diffusion length and promote the bulk transport rate of charge carriers with the consequence of reduced recombination probability. Moreover, the surface atomic geometry and the band structure engineering associated with few-layer-thick g-C3N4NSs will also contribute a lot, as has been primarily demonstrated in several photocatalysts such as ZnO and TiO2.35,36
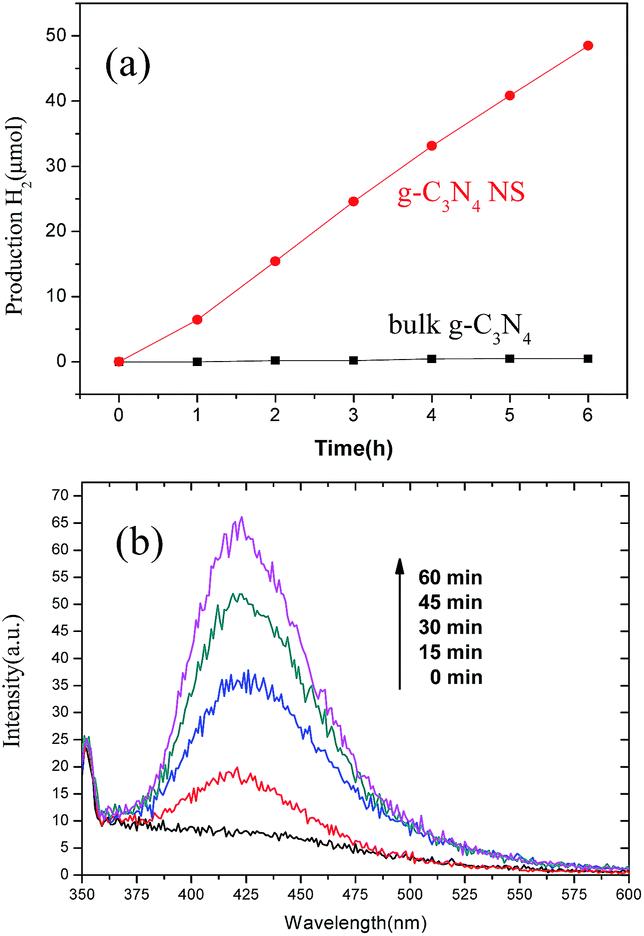 | ||
| Fig. 8 (a) Time-dependent H2 production of bulk g-C3N4 (black curve) and g-C3N4 NSs (red curve) under irradiation with visible light (λ > 400 nm). (b) Fluorescence spectra of 2-hydroxyterephthalic acid (TAOH) solution generated by g-C3N4 NS under λ > 400 nm. | ||
4. Conclusions
In summary, we developed a simple, highly efficient, and large-scale synthesis method to fabricate g-C3N4 NSs and other 2D-layered materials such as MoS2 and WS2 NSs from their parent (bulk) materials. The synthetic process was well controlled to achieve few-layer-thick NSs, and the reaction progress could be visualized by the clear color fading from deep blue to colorless without destroying its entire crystal structure. The enhanced photocatalytic performance of g-C3N4 NSs than its bulk counterpart was attributed to the increased surface area, abundant dangling bonds, and modified electronic structures. This novel route is advantageous over others in terms of both high yield (∼85%) and large-scale synthesis (∼10 g in one time), representing a promising alternative to the current methods in practical applications such as large-scale production of NSs.Acknowledgements
This work was supported financially by the Science Fund for Creative Research Groups of the National Natural Science Foundation of China (grant no. 10821201) and the National Natural Science Foundation of China (grant no. 21103131, 50902037, 51172055, 51372056, and 51372027), the fundamental Research Funds for the Central University (Grant no. HIT.BRETIII.201220, HIT, and NSRIF.2012045, HIT.ICRST.2010008), the Foundation of the National Key Laboratory of Science and Technology on Advanced Composite in Special Environment in HIT, the International Science & Technology Cooperation Program of China (2012DFR50020) and the Program for New Century Excellent Talents in University (NCET-13-0174). CLS is supported by the NSERC, NRC, CIHR, and the University of Saskatchewan.Notes and references
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- K. P. Loh, Q. Bao, G. Eda and M. Chhowalla, Nat. Chem., 2010, 2, 1015–1024 CrossRef CAS PubMed.
- H. S. S. Ramakrishna Matte, A. Gomathi, A. K. Manna, D. J. Late, R. Datta, S. K. Pati and C. N. R. Rao, Angew. Chem., Int. Ed., 2010, 49, 4059–4062 CrossRef PubMed.
- K. Zhang, L. L. Zhang, X. S. Zhao and J. S. Wu, Chem. Mater., 2010, 22, 1392–1401 CrossRef CAS.
- M. J. Allen, V. C. Tung and R. B. Kaner, Chem. Rev., 2010, 110, 132–145 CrossRef CAS PubMed.
- A. V. Murugan, T. Muraliganth and A. Manthiram, Chem. Mater., 2009, 21, 5004–5006 CrossRef CAS.
- K. S. Novoselov, D. Jiang, F. Schedin, T. J. Booth, V. V. Khotkevich, S. V. Morozov and A. K. Geim, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10451–10453 CrossRef CAS PubMed.
- L. H. Li, Y. Chen, G. Behan, H. Z. Zhang, M. Petravic and A. M. Gloshenkov, J. Mater. Chem., 2011, 21, 11862–11866 RSC.
- J. N. Coleman, M. Lotya, A. O'Neill, S. D. Bergin, P. J. King, U. Khan, K. Young, A. Gaucher, S. De, R. J. Smith, I. V. Shvets, S. K. Arora, G. Stanton, H. Y. Kim, K. Lee, G. T. Kim, G. S. Duesberg, T. Hallam, J. J. Boland, J. J. Wang, J. F. Donegan, J. C. Grunlan, G. Moriarty, A. Shmeliov, R. J. Nicholls, J. M. Perkins, E. M. Grieveson, K. Theuwissen, D. W. McComb, P. D. Nellist and V. Nicolosi, Science, 2011, 331, 568–571 CrossRef CAS PubMed.
- Z. Y. Zeng, Z. Y. Yin, X. Huang, H. Li, Q. Y. He, G. Lu, F. Boey and H. Zhang, Angew. Chem., Int. Ed., 2011, 50, 11093–11097 CrossRef CAS PubMed.
- Y. Muramatsu, Y. Tani, Y. Aoi, E. Kamijo, T. Kaneyoshi, M. Motoyama, J. J. Delaunay, T. Hayashi, M. M. Grush, T. A. Callcott, D. L. Ederer, C. Heske, J. H. Underwood and R. C. C. Perera, Jpn. J. Appl. Phys., 1999, 38, 5143–5147 CrossRef CAS.
- P. Niu, L. L. Zhang, G. Liu and H. M. Cheng, Adv. Funct. Mater., 2012, 22, 4763–4770 CrossRef CAS.
- X. D. Zhang, X. Xie, H. Wang, J. J. Zhang, B. C. Pan and Y. Xie, J. Am. Chem. Soc., 2013, 135, 18–21 CrossRef CAS PubMed.
- S. B. Yang, Y. J. Gong, J. S. Zhang, L. Zhan, L. L. Ma, Z. Y. Fang, R. Vajtai, X. C. Wang and P. M. Ajayan, Adv. Mater., 2013, 25, 2452–2456 CrossRef CAS PubMed.
- X. C. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- J. S. Zhang, X. F. Chen, K. Takanabe, K. Maeda, K. Domen, J. D. Epping, X. Z. Fu, M. Antonietti and X. C. Wang, Angew. Chem., Int. Ed., 2010, 49, 441–444 CrossRef CAS PubMed.
- X. C. Wang, K. Maeda, X. F. Chen, K. Takabe, K. Domen, Y. D. Hou, X. Z. Fu and M. Antonietti, J. Am. Chem. Soc., 2009, 131, 1680–1681 CrossRef CAS PubMed.
- Y. D. Hou, A. B. Laursen, J. S. Zhang, G. G. Zhang, Y. S. Zhu, X. C. Wang, S. Dahl and I. Chorkendorff, Angew. Chem., Int. Ed., 2013, 52, 3621–3625 CrossRef CAS PubMed.
- S. Chu, Y. Wang, Y. Guo, J. Y. Feng, W. J. Luo, X. X. Fan and Z. G. Zou, ACS Catal., 2013, 3, 912–919 CrossRef CAS.
- Y. J. Cui, Z. X. Ding, X. Z. Fu and X. C. Wang, Angew. Chem., Int. Ed., 2012, 51, 11814–11818 CrossRef CAS PubMed.
- Z. Z. Lin and X. C. Wang, Angew. Chem., Int. Ed., 2013, 52, 1735–1378 CrossRef CAS PubMed.
- J. G. Zhou, X. T. Zhou, R. Y. Li, X. L. Sun, Z. F. Ding, J. Cutler and T. K. Sham, Chem. Phys. Lett., 2009, 474, 320–324 CrossRef CAS PubMed.
- I. Jiménez, W. M. Tong, D. K. Shuh, B. C. Holloway, M. A. Kelly, P. Pianetta, L. J. Terminello and F. Himpsel, J. Appl. Phys. Lett., 1999, 74, 2620 CrossRef PubMed.
- N. Ferralis, R. Maboudian and C. Carraro, Phys. Rev. Lett., 2008, 101, 156801 CrossRef.
- G. Gui, J. Li and J. X. Zhong, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 075435 CrossRef.
- Z. F. Ding, S. K. Bux, D. J. King, F. L. Chang, H. T. Chen, S. C. Huang and R. B. Kaner, J. Mater. Chem., 2009, 19, 2588–2592 RSC.
- Z. F. Ding, L. Viculis, J. Nakawatase and R. B. Kaner, Adv. Mater., 2001, 13, 797–800 CrossRef CAS.
- R. B. Somoano and A. Rembaum, Phys. Rev. Lett., 1971, 27, 402 CrossRef CAS.
- T. P. Ying, X. L. Chen, G. Wang, S. F. Jin, T. T. Zhou, X. F. Lai, H. Zhang and W. Y. Wang, Sci. Rep., 2012, 2, 426 CAS.
- T. P. Ying, X. L. Chen, G. Wang, S. F. Jin, T. T. Zhou, X. F. Lai, H. Zhang, S. J. Shen and W. Y. Wang, J. Am. Chem. Soc., 2013, 135, 2951–2954 CrossRef CAS PubMed.
- R. R. Dewald, J. Phys. Chem., 1975, 79, 3044–3049 CrossRef CAS.
- J. W. Huffman and W. W. McWhorter, J. Org. Chem., 1979, 44, 594–599 CrossRef CAS.
- J. A. Kerr, Chem. Rev., 1966, 66, 465–500 CrossRef CAS.
- H. B. Feng, R. Cheng, X. Zhao, X. F. Duan and J. H. Li, Nat. Commun., 2013, 4, 1539 CrossRef PubMed.
- H. B. Lu, S. M. Wang, L. Zhao, J. C. Li, B. H. Dong and Z. X. Xu, J. Mater. Chem., 2011, 21, 4228–4234 RSC.
- G. Liu, L. Z. Wang, C. H. Sun, Z. G. Chen, X. X. Yan, L. N. Cheng, H. M. Cheng and G. Q. Lu, Chem. Commun., 2009, 11, 1383–1385 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Equipment and the typical experimental process of LA-assisted lithiation. Dispersion of g-C3N4 NSs after storing for one month under ambient conditions. TEM images and EDS spectra of g-C3N4 NSs. PL spectra of ultrathin g-C3N4 NSs in solution excited at diverse wavelengths and at 340 nm with different excitation intensities. XRD patterns of bulk g-C3N4 before and after soaked in LA. XPS spectra of bulk g-C3N4, and g-C3N4 NSs. Elemental composition before and after the LA-assisted Li intercalation. See DOI: 10.1039/c4ra06036a |
| This journal is © The Royal Society of Chemistry 2014 |