Role of size of drug delivery carriers for pulmonary and intravenous administration with emphasis on cancer therapeutics and lung-targeted drug delivery
Chetna Dhand
a,
Molamma P. Prabhakaran
b,
Roger W. Beuerman
ac,
R. Lakshminarayanan
ac,
Neeraj Dwivedi
d and
Seeram Ramakrishna
*be
aAnti-Infectives Research Group, Singapore Eye Research Institute, Singapore 168751, Singapore
bCenter for Nanofibers and Nanotechnology, Nanoscience and Nanotechnology Initiative, Faculty of Engineering, National University of Singapore, Singapore 117576. E-mail: seeram@nus.edu.sg; Fax: +65-68725563; Tel: +65-65162216
cDuke-NUS SRP Neuroscience and Behavioral Disorders, Singapore 169857, Singapore
dDepartment of Electrical and Computer Engineering, National University of Singapore, Singapore117576, Singapore
eDepartment of Mechanical Engineering, National University of Singapore, Singapore117576
First published on 7th July 2014
Abstract
The design of a drug delivery system and the fabrication of efficient, successful, and targeted drug carriers are two separate issues that require slightly different design parameters. The geometry of the drug carrier such as its size and shape, chemical structure, surface chemistry, and surface charge are among the key parameters that need to be optimized to achieve the desired therapeutic behaviour. We review here the effects of the size of the drug delivery carrier on its biodistribution, target specificity, body clearance rate and, most importantly, the therapeutic action of the drug. Pulmonary and intravenous drug administration are the main focus, with special emphasis on cancer therapeutics and lung-targeted drug delivery. The significance of the effect of the dimensional variations and size of the drug delivery carriers on appropriate controlled and targeted drug delivery is explored with the aim of both prohibiting excessive therapeutic loss via different clearance routes and overcoming side-effects and toxicity.
 Chetna Dhand | Chetna Dhand has completed her PhD in Chemistry from University of Delhi in collaboration with National Physical Laboratory, New Delhi, India. Currently, she is working as a Postdoctoral Research Fellow at Singapore Eye Research Institute, Singapore. Her major research interests include nanomaterials for biomedical applications, targeted drug delivery and biosensors. |
 Molamma P. Prabhakaran | Molamma P. Prabhakaran obtained her PhD in Chemistry from the National University of Singapore (NUS). Currently, she hold an appointment with the Nanoscience and Nanotechnology Initiative in NUS, as a senior research fellow. Her research interests include nanofibers, nanoparticles, tissue engineering and drug delivery. |
 Roger W. Beuerman | Roger Beuerman is currently Senior Scientific Director of the Singapore Eye Research Institute, Professor of Neuroscience and Behavioral Disorders at DUKE-NUS School of Medicine and Adjunct Professor of Ophthalmology, Yong Loo Lin, School of Medicine at the National University of Singapore, and Head of Translational Research, and National Distinguished Professor (FidiPro) of Ophthalmology at the University of Tampere Medical School in Finland. The focus of his work has been in two areas: 1-understanding eye disease for the development therapeutics of infectious disease, and 2-discovery of molecular biomarkers of eye disease for clinical application. |
 R. Lakshminarayanan | Dr R. Lakshminarayanan obtained his PhD from the Department of Chemistry at the National University of Singapore. He was a recipient of the Singapore Millennium Foundation Postdoctoral Fellow and then worked as Research Associate at the University of Southern California. Currently, he is working as a Principal Research Scientist at the Singapore Eye Research Institute. His major interests include antimicrobial peptides design, topical delivery of antimicrobial peptides and protein aggregation diseases. |
 Neeraj Dwivedi | Neeraj Dwivedi obtained his PhD in Physics from the Indian Institute of Technology Delhi, India. Currently, he is working as a Postdoctoral Research Fellow in the Department of Electrical and Computer Engineering, National University of Singapore, Singapore. His major interests include material science, thin films and coatings, MEMS, nanomaterials for biomedical applications. |
 Seeram Ramakrishna | Seeram Ramakrishna, FREng, is the Director of Center for Nanofibers & Nanotechnology at National University of Singapore. He authored 6 books and ∼600 ISI listed journal papers, which attracted ∼35 000 citations and 86 H-index. He is a highly Cited Researcher in Materials Science. He received several awards and recognitions. He is an elected international fellow of Royal Academy of Engineering, UK; National Academy of Engineering, India; Institution of Engineers Singapore; ASEAN Academy of Engineering & Technology; American Association of the Advancement of Science; ASM International; American Society for Mechanical Engineers; Institution of Mechanical Engineers, UK; Institute of Materials, Minerals & Mining, UK; and American Institute for Medical & Biological Engineering. He is an editorial board member of ∼ 10 international journals. |
1. Introduction
A drug delivery system (DDS) is defined as the mechanism or strategy used to introduce a therapeutic agent into the body.1 The most important challenge in transferring any DDS to a clinical setting is to identify the optimum physicochemical parameters that simultaneously determine molecular targeting, immune evasion, and controlled drug release.2 This depends on the complex interdependence of the properties of the DDS (its composition, size, shape, surface charge, hydrophilicity, and the type and density of the ligand), the properties of the payload (the type of drug and its solubility, loading, and release kinetics), and in vivo physiological barriers to trafficking of the DDS (immune surveillance, particle extravasation, tissue penetration, and cellular uptake).3 Delivering the appropriate amount of the desired drug to the target organ or area without causing any side-effects while also preventing the induction of drug resistance is a daunting task, but is an important requirement in a targeted DDS.The commercialization of nanotechnology in pharmaceutical and medical science has revolutionized this field and has led to a new era of nanomedicines.4,5 Highly efficient DDSs based on nanovehicles could potentially reduce the drug dose needed to achieve a therapeutic benefit, which, in turn, may lower the cost and/or reduce the side-effects associated with particular drugs. Furthermore, the size, shape, and surface characteristics of nanoparticles can be easily manipulated to achieve both passive and active drug targeting. Drug delivery carriers based on different nanoparticles have emerged in the past few decades, e.g. nanocrystals, liposomes, polymer micelles, dendrimers, and polymer–drug conjugates.4–12 Table 1 gives a detailed description of various currently available commercial drugs based on nanocarriers.
| Type of targeted drug delivery carrier | Commercial drug | |||||
|---|---|---|---|---|---|---|
| Brand name | Active ingredient | Year of approval and license holder | Indication | Mode of administration | Ref. | |
Nanocrystal: nanoscale formulation of the drug itself with outer thin coating/layer of non-ionic surfactant or polymeric macromolecule. It can function as its own carrier |
Rapamune | Sirolimus | 2001, Wyeth | Immunosuppression | Oral | 4 and 6 |
| Emend | Aprepitant | 2003, Merck | Anti-emetic | Oral | 4 and 6 | |
| Tricor | Fenofibrate | 2004, Fournier and Abbott | Hypercholesterolemia | Oral | 4 and 6 | |
| Megace | Megestrol | 2005, Elan/Par Pharm | Anti-anorexia | Oral | 4 and 6 | |
Liposomes (30 nm–20 mm): liposomes are self-assembled artificial vesicles developed from amphiphilic phospholipids. Liposomes are the most clinically established nanosystems for drug delivery due to their ability to entrap both hydrophilic and hydrophobic drugs |
AmBsome, 80 nm | Amphotericin B | 1995, Glead | Severe fungal infections | Intravenous | 7 and 8 |
| Depocyt, 200–300 nm | Cytarabine | 2002, Napp | Lymphomatous meningitis | Spinal | 7 and 8 | |
| Doxil and Caelyx, 85 nm | Doxorubicin | 1995, Schering-Plough | Ovarian cancer, Kaposi's sarcoma and breast cancer | Intravenous | 7 and 8 | |
| Daunoxome, 45 nm | Daunorubicin | 1996, Diatos | Blood cancer | Intravenous | 7 and 8 | |
| Visudyne | Verteporfin | 2000, Novartis AG/QLT | Age-related macular degeneration | Intravenous | 7 and 8 | |
Polymer–drug conjugate (6–15 nm): in polymer–drug conjugates the drug molecules are bound to macromolecular structures to enhance their circulation time in blood and to increase their solubility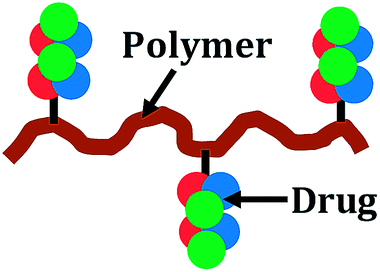 |
Adagen | Adenosine deaminase | 1993, Enzon | Immunodeficiency disease | Intramuscular | 9 and 10 |
| Oncaspar | L-Asparaginase | 1997, Enzon | Antineoplastic | Intravenous, intramuscular | 9 and 10 | |
| Neulasta | Pegfilginase | 2002, Amgen | Reduction of febrile neutropenia associated with chemotherapy | Subcutaneous injection | 9 and 10 | |
| PEG-Intron | Interferon α-2b | 2001, Enzon, Schering-Plough | Hepatitis C | Subcutaneous | 9 and 10 | |
Polymeric micelles (20–150 nm): polymeric micelles are self-assembled core–shell nanostructures formed in aqueous solution consisting of amphiphilic block copolymers. Polymeric micelles have the advantage over other nanocarriers of having a very small size, which is very important for percutaneous lymphatic delivery or extravagation from blood vessels into the tumor tissue. These micelles also have a large loading capacity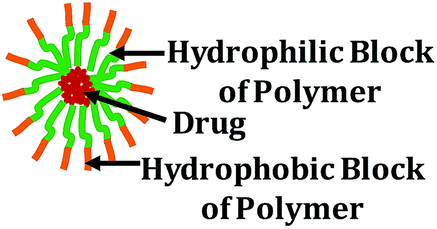 |
Genexol-PM | Paclitaxel | 2007, Samyang biopharmaceuticals | Cancer chemotherapy | Intravenous | 4 |
Dendrimers (5–10 nm): dendrimers have novel three-dimensional, hyperbranched globular nanopolymeric architectures. Characteristics such as nanoscopic size, a narrow polydispersity index, excellent control over molecular structure, availability of multiple functional groups at the periphery and cavities in the interior make them suitable and excellent candidates for targeted drug delivery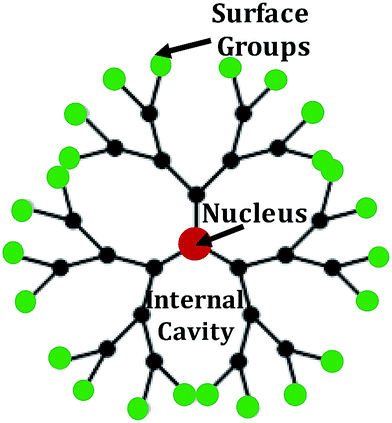 |
VivaGel, 3–10 nm | SPL7013, dendrimer | 2014, Starpharma | Bacterial vaginosis or vaginal microbicide for prevention of HIV and HSV infections | Topical | 11 |
| Protein (albumin) nanoparticles (130 nm): | Albraxane, 130 nm | Paclitaxel | Metastatic breast cancer | 2005, Abraxis biosciences | Intravenous | 12 |
| Lipid colloidal dispersion (100–150 nm): | Amphotec, 122 ± 48 nm | Amphotericin B | Fungal infections | 1996, Sequus pharmaceuticals | Intravenous | 13 |
Physicochemical properties such as the size, shape, and surface characteristics of a DDS are among the key parameters which should be considered from an engineering perspective when designing an efficient DDS for a particular therapeutic agent via a specific administration route. Venkataraman et al.14 and Champion et al.15 have previously published detailed reviews of the effect of the shape of drug delivery nano- or microcarriers on their therapeutic performance. In addition, Honary and Zahir16,17 published a detailed two-part review on the effect of the zeta potential and surface charge of nanocarriers on their drug delivery characteristics. However, no review has yet been published compiling the effect of the size of the drug carrier on its drug delivery characteristics. The size and dimensions of the drug delivery carrier influence almost every aspect of its functioning and efficacy, such as degradation, flow properties, renal clearance, hepatic filtration, tissue extravasation/diffusion, and endocytosis.18 For example, nanoparticles <10 nm can be easily cleared by excretion through the kidneys, whereas larger nanoparticles (>150 nm) are more likely to be removed by the mononuclear phagocyte system (MPS), also known as the reticuloendothelial system.19 It has been shown that liposomes 100–150 nm in size have a higher potential to be part of the blood circulation for a longer time and also show higher hepatic filtration kinetics than liposomes >70 nm.20,21 The size of the DDS also plays a vital role in determining accumulation in the region of the tumor through the enhanced permeability and retention (EPR) effect,22 which is discussed in detail later in this review. As a physical parameter, the size of a DDS is critical with respect to the drug target or site of action, the cellular uptake or internalization mechanism, and specialized therapeutic action.23
We highlight here the importance of the size of the drug delivery carrier in deciding its biodistribution, target accumulation or specificity, body clearance rate, and, most importantly, its therapeutic action. We focus on two different modes of therapeutic administration: pulmonary administration (targeting the lungs or other organs via the pulmonary route or human respiratory tract) and intravenous administration. We place special emphasis on cancer therapeutics and lung-targeted intravenous administration (targeting the lungs via the blood circulation).
2. Effect of size of drug delivery systems on pulmonary administration
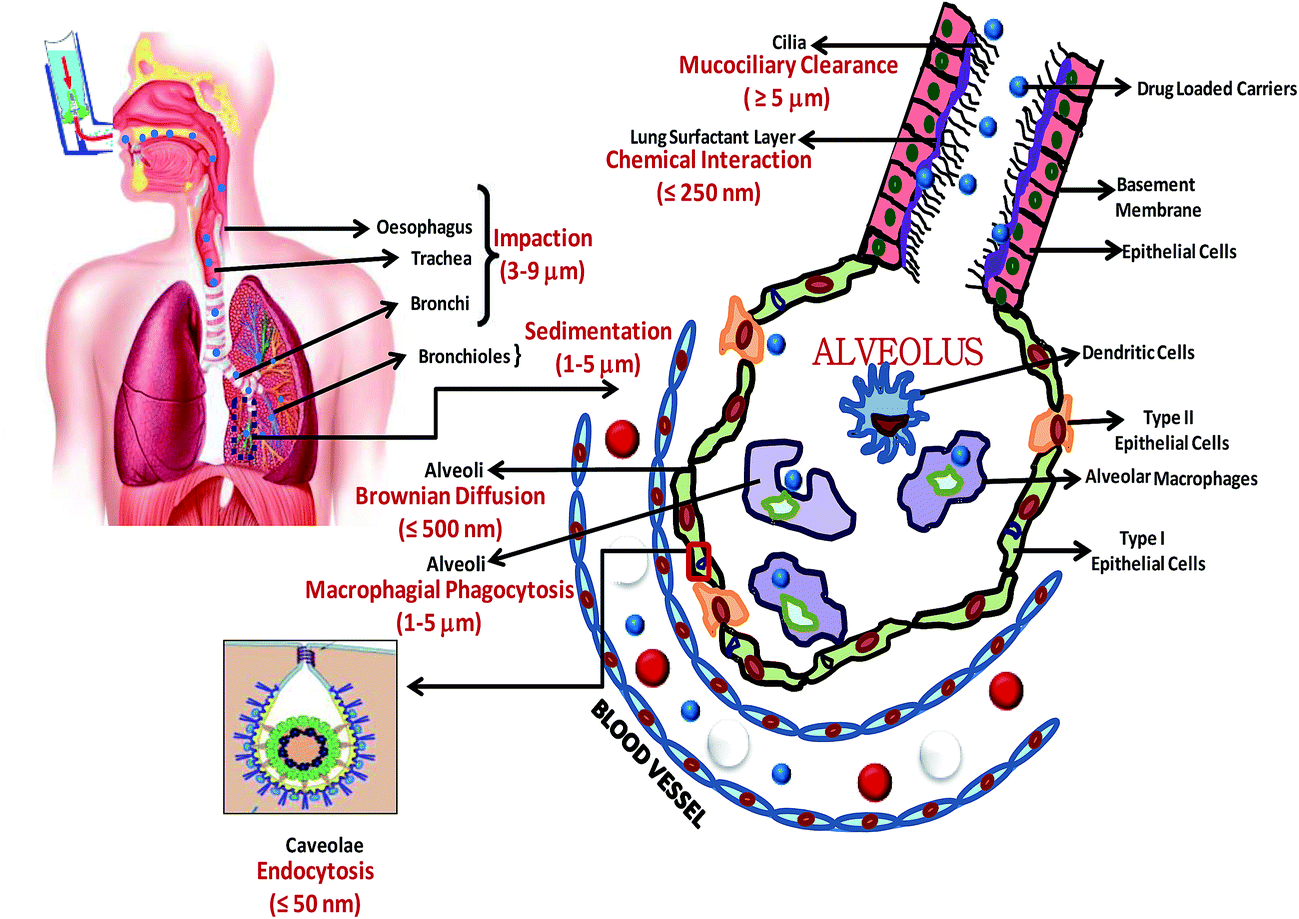
The physiology of the lung makes it an ideal target organ for drug delivery. The pulmonary route serves as an appropriate mode of administration due to its high solute permeability, large surface area for absorption with non-invasive characteristics, and the fact that it is a site with limited proteolytic activity.24 During pulmonary administration, drugs are delivered locally into the lungs for the treatment of respiratory diseases such as asthma and lung cancer; which has the potential to reduce dose-dependent drug toxicity.25 Systemic drug delivery can also be achieved by targeting the drug delivery carriers to the alveolar region, where the drug can be absorbed through the thin epithelial cell layer to enter the systemic circulation.26,27 This may be desirable to achieve a rapid onset of action by avoiding first-pass metabolism and for delivering biotherapeutic agents, e.g. peptides and proteins, that cannot be delivered orally as a result of enzymatic degradation and the poor permeability of the intestinal membrane.28 The lungs can also be targeted for the delivery of drugs to specific lung cells, such as alveolar macrophages, for the treatment of diseases such as tuberculosis.29The human respiratory or pulmonary system is divided into two main functional zones: the conducting zone (consisting of the trachea, bronchi, and bronchioles) and the respiratory zone (consisting of the airways and the alveoli). The human lung contains about 2300 km of airways and 500 million alveoli, which actively participate in the gaseous exchange process.30 The surface area of the human lungs is estimated to be approximately 75–140 m2 in adults.31 The pseudostratified epithelium, which provides a barrier for absorption into the bloodstream, is different in different parts of the lungs. The airway epithelia are composed of gradually thinning columnar epithelium and the bronchial and bronchiolar epithelia have thicknesses of 3.5 mm and 0.5–1 mm, respectively.24 In contrast, the alveoli epithelium is only a single cell thick and the distance from the alveolar lumen to the bloodstream is <400 nm. The large accessible surface area of the alveoli and the intimate air–blood contact in this region makes this zone a suitable site for gaseous exchange as well as for the absorption of inhaled aerosols, including drug delivery nanovectors or nanomedicines.32 Pulmonary DDSs are based on the principle of aerosolization. Aerosols containing uniformly sized particles organized with drug-loaded vehicles may deliver a uniform drug dose and have uniform drug release kinetics.33
The location and extent of drug carrier deposition and the efficiency of the drug-loaded nanovehicle after inhalation is strongly influenced by three main factors: the size and geometry of the DDS; the anatomy of the upper and lower airways with an alveolar structure; and the ventilatory parameters. The ventilatory parameters include the breath pattern (i.e. breath-holding and the presence of expiratory flow limitation), flow-rates, and the tidal volume, which together determine the airflow velocity and the residence time in the respiratory tract.34 Depending on the size of the drug delivery vector, there are again three principal mechanisms that decide its deposition and distribution in the lungs: inertial impaction; gravitational sedimentation; and Brownian diffusion (Table 2).35 Deposition generally refers to the mean probability of a particle being deposited in the respiratory tract when settling on the airway surfaces. For lung deposition, the particle size is characterized by the mass median aerodynamic diameter (Da), which is the diameter of a spherical particle with a density of 1 g cm−3 with the same settling velocity (under gravity through air) as the particle of interest. This is given by the relation:36
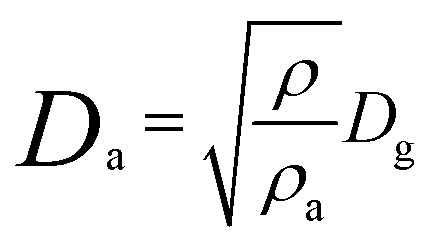 | (1) |
| Size of inhaled aerosol particles (μm) | Site of particle deposition | Mechanism of deposition |
|---|---|---|
| 5–9 (slow inhalation) | Large airways, including oropharynx, trachea, and bronchi | Inertial impaction |
| 3–6 (fast inhalation) | Large airways, including trachea and bronchi | Inertial impaction |
| 1–5 | Smaller airways | Gravitational sedimentation |
| ≤0.5 | Alveoli | Brownian diffusion |
A widely accepted theory states that for efficient lung deposition, the aerodynamic diameter should be in the range 1–5 μm.37,38 Small particles (<1 μm) are likely to be absorbed quickly from the airways and this poses a risk of systemic toxicity. Moreover, 80% of administered particles <1 μm are exhaled without being deposited due to their low inertia.39 In contrast, large particles are cleared by the mucociliary clearance mechanism.40 Some studies have suggested that particles with a density <0.4 g ml−1 and a geometric diameter >5 μm are deposited efficiently in the lungs.41 Therefore, to successfully arrive at the deeper lung tissues, the inhaled particles should be small enough to avoid deposition in the upper airways by sedimentation or impaction and at the same time should be large enough to avoid loss by exhalation. Therefore a particle size in the range 1–5 μm is required to achieve efficient pulmonary drug delivery. Hirota et al.42 studied the distribution and deposition of respirable PLGA microspheres with incorporated drugs for tuberculosis (coumarin-6 and rifampicin) with diameters of 2.67 ± 2.18 μm (for coumarin-6) and 2.35 ± 1.96 μm (for rifampicin) in small animal models. Their results showed a maximum accumulation of microspheres in the tracheal and primary bronchi regions. PLGA microspheres ∼3 μm in diameter were suggested as the most suitable size for phagocytic uptake by alveolar macrophages.43
Drug-carrying vectors are absorbed via different biological routes in the respiratory tract depending on their size. In the upper conducting region, nanomedicines loaded with drugs start to deposit into the mucous layer (∼5 μm deep and composed of electrolytes, proteins, glycoproteins, and cell debris) that lines the airways, or the surfactant layer (10–20 nm thick, 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 wt% phospholipids and specific proteins) covering the alveolar region.44 The nature of the drug delivery vehicle does not play a role in determining the extent of submergence of guest nanocarriers into lining fluids after deposition; the size of the nanovector is the determining factor. Stuart et al.45 studied the interaction of the surfactant film in the lungs with nanoparticles of two different sizes (187 and 230 nm) and indicated that the extent of nanoparticle incorporation into the surfactant layer depends on the dimensions of the nanoparticles, particularly their size. The results of their study show a stronger interaction between the smaller nanoparticles and the surfactant monomolecular film than with larger nanoparticles. The smaller the size of the nanoparticles, the greater is the absorption of these nanovehicles into the surfactant layer. After inclusion in the fluid lining the lung, there are separate biokinetic mechanisms for lung absorption and non-absorptive clearance.46 Most of the small and highly soluble hydrophobic molecules undergo rapid absorption through the lung epithelial membrane by passive diffusion.47 The kinetics of passive diffusion in the alveoli is much faster than in the smaller airways because most pulmonary absorption occurs through the alveolar capillaries of the alveolar region. A smaller portion of the inhaled nanoparticles are absorbed from the tracheobronchial airways.34 In contrast, low molecular weight hydrophilic molecules are absorbed by active transport processes depending on regional expression in the lung and the functionality of the receptors or transporters. Bitonti et al.48 reported the absorption of large immunoglobulin (IgG) molecules in the upper airways by receptor-mediated transcytosis of IgG.
:1 wt% phospholipids and specific proteins) covering the alveolar region.44 The nature of the drug delivery vehicle does not play a role in determining the extent of submergence of guest nanocarriers into lining fluids after deposition; the size of the nanovector is the determining factor. Stuart et al.45 studied the interaction of the surfactant film in the lungs with nanoparticles of two different sizes (187 and 230 nm) and indicated that the extent of nanoparticle incorporation into the surfactant layer depends on the dimensions of the nanoparticles, particularly their size. The results of their study show a stronger interaction between the smaller nanoparticles and the surfactant monomolecular film than with larger nanoparticles. The smaller the size of the nanoparticles, the greater is the absorption of these nanovehicles into the surfactant layer. After inclusion in the fluid lining the lung, there are separate biokinetic mechanisms for lung absorption and non-absorptive clearance.46 Most of the small and highly soluble hydrophobic molecules undergo rapid absorption through the lung epithelial membrane by passive diffusion.47 The kinetics of passive diffusion in the alveoli is much faster than in the smaller airways because most pulmonary absorption occurs through the alveolar capillaries of the alveolar region. A smaller portion of the inhaled nanoparticles are absorbed from the tracheobronchial airways.34 In contrast, low molecular weight hydrophilic molecules are absorbed by active transport processes depending on regional expression in the lung and the functionality of the receptors or transporters. Bitonti et al.48 reported the absorption of large immunoglobulin (IgG) molecules in the upper airways by receptor-mediated transcytosis of IgG.
For nanovectors that are completely insoluble in mucus and the lining fluids, there are different post-defense mechanisms available in the body for the removal of deposited nanoparticles and maintenance of the lung mucosal surface, including mucociliary escalator transport, phagocytosis by macrophages, and endocytosis.49 The mucociliary escalator dominates the clearance of comparatively larger particles from the upper airways by the action of ciliated epithelial cells pushing the mucus and the particles deposited on the airway walls to the larynx, where they are swallowed into the gastrointestinal tract or excreted through the mouth.50 These deposited structures may also be removed by coughing within 1–2 days of administration.24
Macrophage phagocytosis and endocytosis are the main modes of clearance for slowly dissolving and insoluble nanoparticles from the alveolar region.51 There are around 500 million alveoli in the lungs and these are consistently examined on the air-side surface by 12–14 alveolar macrophages in the lung lining fluid.29 The particle size plays an important role here in deciding the uptake of deposited particles by alveolar macrophages. Particles 1–3 μm in diameter are taken up by macrophages (with a cell diameter of 15–22 μm) better than those 6 μm in diameter.52 Particles <0.26 μm may escape phagocytosis.53 These small nanoparticles will interact further with non-phagocytic cells of the epithelium and initiate endocytic events regulated by clathrin-coated pits and caveolae.54 Caveolae are indentations in the plasma membrane lined with caveolin-1 and are predominantly expressed by lung capillaries and type I alveolar cells. Particles of several nanometers in radius may be transported within caveolae from the lung to the bloodstream.55 Inspiratory expansion and expiratory contraction of the lung alveoli may lead to opening and closing of the caveolae. These openings measure between 40 and 100 nm in size and are thought to be involved in the transport of macromolecules, such as proteins, across the alveolar-capillary barrier.56
These processes of phagocytosis by macrophages and/or endocytosis by the epithelial and endothelial cells will result in extrapulmonary nanovector translocation to various sites depending on their size, chemical composition, particle size, surface characteristics, labelling materials, and the experimental model.57 Rapid and excessive translocation of 13C-labelled nanoparticles with diameters of 26 nm have been reported in the liver within 1 day of administration via the pulmonary route in a rat model.58 Kreyling et al.59 estimated the biodistribution of 1% iridium nanoparticles (10–20 nm diameter) after inhalation and the maximum nanoparticle accumulation was observed in the liver, spleen, kidneys, brain, and heart. In a rat model, after 3 months of exposure by inhalation to ultrafine (∼20 nm) and fine (∼200 nm) titanium dioxide particles, the ultrafine particles were cleared significantly more slowly and showed more translocation to interstitial sites and to regional lymph nodes than fine titanium dioxide particles.60 Particles between 20 and 50 nm in diameter may enter the central nervous system and cells. In addition, alveolar macrophages on the surface of the lungs are unable to recognize particles <70 nm as being foreign, thus allowing them to gain access to the pulmonary interstitium and to the capillary blood flow.61 Fig. 1 shows the distribution of drug-loaded carriers in the lungs with respect to size.
 | ||
| Fig. 1 Schematic diagram showing different routes for distribution of pulmonary drug carriers, their clearance and absorption with respect to size dependence and preference. | ||
Some of the FDA-approved inhaled formulations and vaccines administered via the pulmonary route include the following (all these drugs fall in the narrow size range 1–10 μm): NebuPent (drug, pentamidine isothionate; size, 1–2 μm; treatment for pneumocystis carinii pneumonia); Virazole (drug, ribavirin; size, <1 μm; RSV lower respiratory tract infection); Tobramycin Inhalation Solution (TOBI; drug, tobramycin; size, 0.5–10 μm; first inhaled antibiotic given by nebulizer); Amphotericin B Inhalation Powder (ABIP; size, 1–5 μm; inhaled antifungal product); Ventavis (drug, iloprost; size, 1–3 μm; inhaled treatment for pulmonary arterial hypertension); and Resmycin (drug, doxorubicin HCl; size, <2 μm; inhalation solution for lung cancer therapeutics).
In summary, the size of the drug delivery vehicle not only affects its pulmonary distribution, but also its metabolism, clearance, and absorption. With respect to the drug carrier distribution, particles with sizes in the range 5–9 μm are more likely to stay in the upper airway region, including the oropharynx, trachea, and bronchi. Particles with dimensions of 1–5 μm and ≤0.5 μm are more likely to deposit in the smaller airways (bronchi and bronchioles) and terminal alveoli, respectively. With respect to the clearance efficiency, vehicles <1 μm have are greater tendency to be exhaled as a result of low inertia and those >5 μm can be swept out easily as a result of the mucociliary mechanism. In view of the effect of size on the metabolism and absorption of drug-loaded carriers, small (>250 nm) hydrophobic particles show rapid absorption through the lung epithelial membrane by passive transport and similar sized hydrophilic particles undergo active transport via endocytosis. However, larger particles (>1 μm) undergo absorption and translocation by receptor-mediated transcytosis and macrophagial phagocytosis. Particles in the size range 1–3 μm are reported to be most suitable for macrophagial uptake.
3. Effect of size of drug delivery carriers on intravenous administration, with special emphasis on cancer therapeutics and lung-targeted intravenous drug delivery
Intravenous administration is an important drug delivery route, particularly if the target organ is far from the site of administration. As with other modes of therapeutic administration, the size of the drug delivery vehicle is an important parameter in deciding the target and biodistribution of the drug, the route and rate of drug clearance, and, consequently, the desired therapeutic action. During passage through the vascular bed and before reaching the target site, the drug carrier undergoes different biodistribution steps depending on its size. For example, nanoparticles <20–30 nm can be easily cleared from the blood via renal clearance, whereas nanoparticles with dimensions of 30–150 nm are more likely to accumulate in the bone marrow, heart, kidney, and stomach. Nanoparticles >150 nm are generally found in the liver and spleen. Larger nanoparticles can be easily taken up by the MPS. Thus the drug delivery carriers can easily escape from the blood circulation to different body organs or parts through the openings available at the endothelial barrier, also known as fenestrations. To pass this continuous and intact endothelial barrier, the particle/carrier size should be <150 nm under normal conditions. However, under different pathological conditions the vasculature and fenestration size undergo changes, e.g. in cancerous tissues the rapid growth and multiplication of cells demands a higher blood supply that leads to the development of a neovasculature characterized by a discontinuous endothelium with large fenestrations of 200–780 nm. Table 3 summarizes the fenestration sizes of various vital organs in animals.| Organ and animal model | Fenestration size (nm) | Ref. |
|---|---|---|
| Organ: kidney, animal model: rat or guinea pig | 20–30 | 62 |
| Organ: liver, animal model: mouse | 150 | 63 |
| Organ: spleen, animal model: mouse | 150 | 63 |
| Organ: lung, animal model: dog | 1–400 | 64 |
| Organ: bone marrow, animal model: rat or guinea pig | 85–150 | 65 |
| Organ: skeletal, cardiac and smooth muscles, animal model: mouse | ≤6 | 66 |
| Organ: skin, subcutaneous and mucous membrane, animal model: mouse | ≤6 | 66 |
| Organ: blood–brain barrier, animal model: in vitro model | No fenestration | 67 |
| Organ: tumor in ear and brain, animal model: mouse | 200–380 | 68 |
| Organ: tumor in brain, animal model: mouse | 100–380 | 69 |
Joliano et al.70 reported a direct relation between the rate of clearance of encapsulated liposomal vesicles from the bloodstream and their particle size. Attempts have been made to evaluate carrier endocytosis, trafficking, and eventually intracellular fate within endothelial cells lining the vascular lumen with respect to the size (0.1–10 μm) and shape (spheres versus elliptical disks) of intracellular adhesion molecule-1 (ICAM-1) targeted polymer carriers administered intravenously. These size-dependant studies show that the endothelial cells internalized polymer carriers coated with anti-ICAM up to several microns in size via endocytosis mediated by cell adhesion molecules. Micron-sized carriers have also been found to have prolonged residency in pre-lyposomal compartments, whereas submicron carriers are trafficked more readily to liposomes. The rational design of carrier geometry might be helpful in optimizing endothelium-targeted therapeutic agents.71 Koval et al.2 examined the uptake and transport of IgG-opsonized polystyrene beads of particular dimensions ranging from 0.2 to 3 μm using macrophages derived from mouse bone marrow. Although the kinetics of the internalization of the opsonized beads was comparable for different sized particles, the internalization mechanism was shown to be dependent on size. The smaller particles (0.2–0.75 μm) were internalized by clathrin-mediated endocytosis, whereas actin-dependent phagocytosis played an important role in the internalization of larger particles (1–3 μm).
Illum et al.3 systematically studied the blood clearance and subsequent organ deposition profile after intravenous administration of colloidal particles of different sizes, shapes, and nature. Small polystyrene microspheres (1.27 μm) were removed by the retinoendothelial system and were retained by Kupffer cells in the liver. Conversely, large polystyrene particles (15.8 μm) became lodged in the capillary beds of the lungs as they are larger than the critical size for passage through the pulmonary vascular bed.3 Kanke et al.72 have also reported the liver as the primary deposition center for small spherical polystyrene particles <7 μm, whereas particles >7 μm were filtered mechanically and retained for prolonged periods in the lungs. The size of the DDS also influences its splenic and renal clearance profile. It has been reported that particles >200 nm are more prone to elimination via splenic filtration, whereas particles <20 nm are susceptible to clearance through kidneys. Table 4 shows the indirect proportionality relation between the hydrodynamic diameter (HD) of different globular proteins and their biodistribution, with special emphasis on renal filtration (modified version of Table 1 in Choi et al.73).
| HD = A × MWB + C × MWD |
| Protein molecule | Molecular weight (kDa) | Hydrodynamic diameter (nm) | Urine/blood filterability (%) | Blood half-life (min) | Whole-body half-life (min) |
|---|---|---|---|---|---|
| a ScFv: single-chain variable fragment; Fab: portion of antibody; HSA: native albumin; IgG: immunoglobulin. | |||||
| Insulin | 5 | 3.0 | 100 | 9 | 1.9 |
| Myoglobin | 17 | 3.8 | 75 | 9 | 2.0 |
| ScFv | 30 | 5.3 | 74 | 11 | 1.4 |
| Fab | 50 | 6.0 | 9 | 28 | 4.0 |
| HSA | 67 | 7.3 | 0.3 | 110 | 16.0 |
| IgG | 152 | 11.0 | <0.1 | 330 | 730 |
The mammalian vasculature has an average pore size of ∼5 nm and hence a DDS with a size approaching this value shows rapid equilibration between the agents injected intravenously and the extracellular space.73 Above this value, however, transport across the endothelium is extremely slow. For non-biodegradable nanoparticles, other routes for the elimination of the nanoparticle are through the liver, into bile, or into feces. The liver specifically takes up and eliminates nanoparticles with an HD in the range 10–20 nm. To bypass this removal step via the retinoendothelial system, therapeutic agents can be coated with specialized materials such as polyethylene glycol (PEG). Extraction of the nanoparticles into bile is an extremely slow and inefficient step and can be ignored. Particles with dimensions <10 nm can leave the systemic circulation through the permeable vascular epithelium of the lymph nodes. These small particles can be eliminated by following the sinus endothelium route of bone marrow.74 Choi et al.73 reported an interesting study on the intravenous administration of quantum dots (QDs) (with a ZnS shell and CdSe core) of different HDs (4.36, 4.99, 6.70, and 8.65 nm) into the bodies of rats followed by estimation of their clearance from blood, their biodistribution, and body clearance parameters as a function of size. The blood concentration studies carried out at different time intervals showed the following trend: 8.36 > 6.70 > 4.99 > 4.36 nm. This shows that the larger nanoparticles have a greater tendency to remain in the bloodstream and therefore have a longer half-life in blood; this trend was reversed during the urine elimination studies. Four hours after intravenous injection, QDs with an HD of 8.36 nm showed a distribution trend of liver (maximum concentration) > kidney > spleen ≈ intestine > feces (least concentration). This trend was reversed for QDs with an HD of 4.36 nm.
Minchin75 has reported the effect of the size and charge of gold–dendrimer nanoparticles with respect to their biodistribution in mice. A progressive decrease in nanoparticle concentration in the kidney with an increase in particle diameter from 5 to 22 nm was reported, whereas the lungs, liver, and spleen showed a continuous accumulation of the nanoparticles as their size increased. These results clearly show that, without an attached target molecule, these nanoparticles can selectively enter specific organs solely on the basis of their charge and size.
Intravenous drug delivery is undeniably one of the most efficient, rapid, and common routes of administering drugs, but this mode of drug delivery has received special attention in cancer therapeutics. Considering the high impact of research on cancer nanomedicines and the significant effect of the size of drug delivery carriers on the therapeutic performance of chemotherapeutic drugs, we have reviewed the effect of the size of drug delivery nanocarriers administered intravenously in cancer therapeutics. As we have already discussed the effect of size on pulmonary administration earlier in this paper, the effect of the size of drug carriers on lung-targeted drug delivery by the intravenous route is also discussed.
3.1 Effect of size of drug delivery vehicles on cancer therapeutics
Cancer drugs have great potential within the therapeutic market mainly because cancer is the second leading cause of death worldwide after cardiovascular disease. Approximately 12.5 million new cases of cancer are diagnosed worldwide each year and considerable research is in progress for methods of drug delivery in cancer treatment. Cancer drug delivery is no longer about simply wrapping up cancer drugs in new formulations for different routes of delivery. The focus is on targeted cancer treatment. Targeted drug delivery limits side-effects, requires fewer doses, and facilitates the targeting of cancerous tissues while leaving healthy areas of the body unaffected. According to a technical market research report,76 the global market for cancer treatment was worth $47.3 billion in 2008. This was estimated to increase to over $110.6 billion by 2013 with a compound annual growth rate of 12.6%. Of the four main types of cancer treatment (chemotherapy, hormone treatment, target treatment, and immunotherapy), the target treatment segment has the largest share of the market.Advanced cancer therapeutics requires the development of drug delivery carriers with highly specific targeting and enhanced drug bioavailability/loading with few chemotherapeutic side-effects. DDSs accumulate in solid tumors through the EPR effect, characterized by the leaky blood vessels and impaired lymphatic drainage in tumor tissues.22 The size of the DDS plays a crucial role when targeting drugs to cancer cells within the tumors. Anthracyclines are a class of drugs derived from Streptomyces peucetius var. caesius that are commonly used to treat a range of cancers, including breast cancer, lung cancer, stomach cancer, some leukemias, and Hodgkin's lymphoma.77,78 However, anthracyclines, including doxorubicin and daunorubicin, are notorious for causing cardiotoxicity and neutropenia. Particle sizes of around 100 nm are too large to leave healthy blood vessels, but can easily escape through the leaky and hastily built tumor-feeding vasculature. Therefore to decrease these cytotoxic effects by using smaller particles, several liposomal and particulate drug formulations have been designed and investigated as efficient drug delivery nanovectors.
Compared with conventional drugs, the encapsulation of anthracyclines within liposomes significantly alters their pharmacokinetic profiles and promotes selectively high drug concentrations in tumors.79 Conventional liposomes used for drug delivery purposes typically have sizes of <300 nm, are composed of naturally occurring or synthetic phospholipids, and are reported to be easily internalized by MPS cells. They have enormous potential to protect other body tissues from the dose-dependent toxic effects of these drugs.80 Myocet is one such liposome-encapsulated doxorubicin drug, introduced by Elan Pharmaceuticals, USA as a multiple vial kit composed of liposomes ranging from 150 to 250 nm in size, lyophilized doxorubicin, and a citric acid buffer. These individual components are mixed at the point of care and result in highly efficient loading of the therapeutic agent within the liposomes. Myocet does not affect the drug circulation time, but reduces the chances of cardiotoxicity and neutropenia.81
Similarly, Doxil (also known as Caelyx in Europe) is a novel ∼85 nm “stealth” liposomal formulation of doxorubicin in which the liposomes contain surface-grafted segments of the hydrophilic polymer PEG (Fig. 2).82 The smaller size of these nanocarriers equilibrates the drug-carrying capacity and circulation time and allows their extravasation through endothelial defects/gaps in the microvasculature of the tumor.83 These sterically stabilized PEGylated liposomes show reduced interactions with plasma proteins and mononuclear phagocytes and consequently have greatly prolonged circulation times. Doxil has been reported to result in only one-third of the incidence of congestive heart failure compared with conventional doxorubicin, resulting in “a quantum jump in quality of life”.84 It is considered to be the most efficient liposomal drug delivery formulation and has so far achieved the most prolonged circulation, with a terminal half-life of 55 hours in humans with enhanced bioavailability of the drug in the cancer cells. DaunoXome is another nanosized daunorubicin-containing liposomal formulation with a markedly prolonged circulation time and enhanced tumor accumulation. It was designed by Gilead Sciences, Inc. (Forest City, CA, USA) and a series of modifications to the liposome structure helps to retard uptake by mononuclear phagocytes. Its liposomal composition includes a lipid bilayer of distearoylphosphatidylcholine and cholesterol in a 2:1 molar ratio. This liposomal daunorubicin provides an extended circulation time due to its small size (∼45 nm) and rigid bilayer; it is highly efficient against Kaposi's sarcoma and other tumors.85
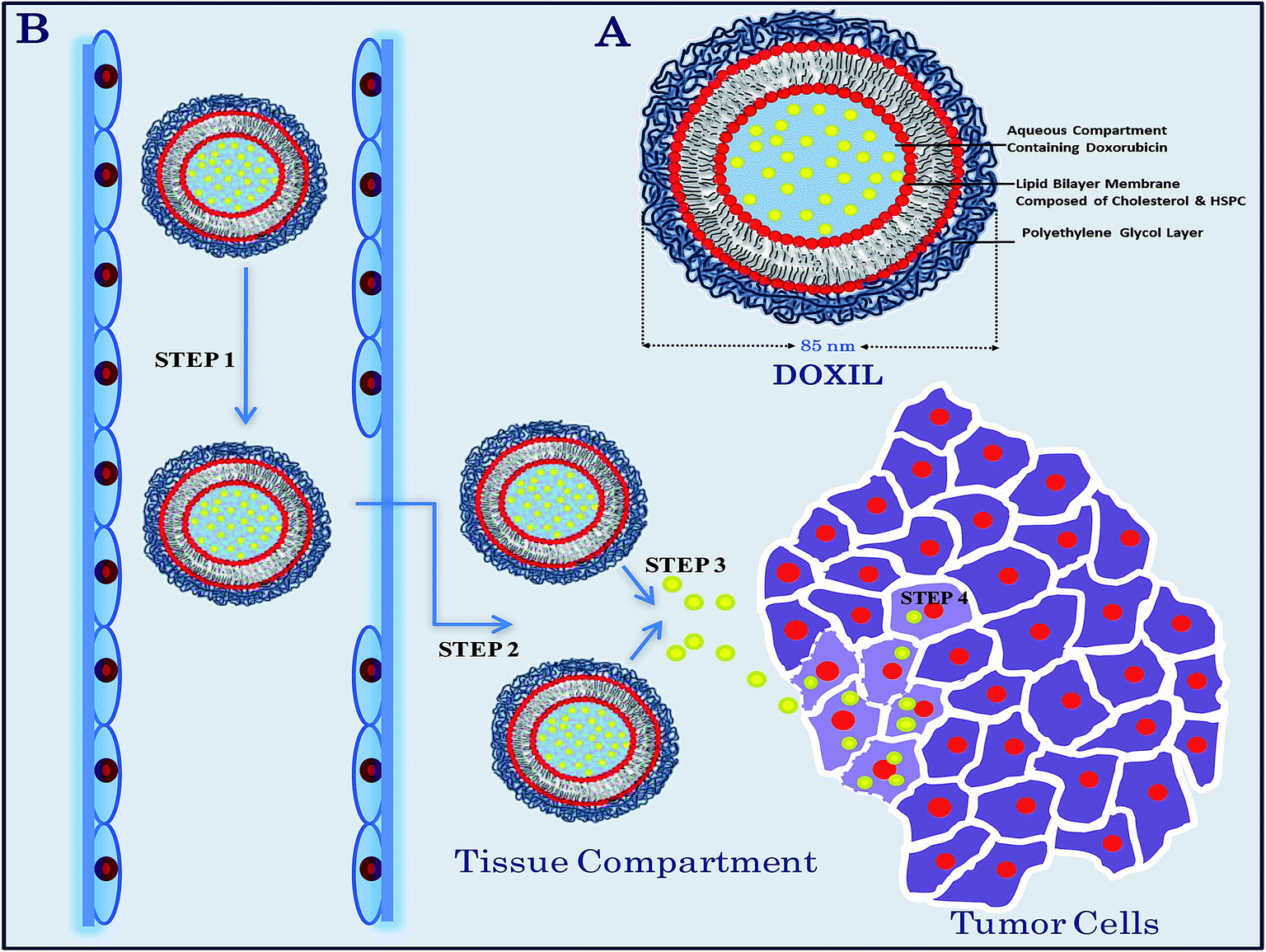 | ||
| Fig. 2 (A) Chemical structure of a Doxil liposome. (B) Schematic diagram showing the proposed mechanism of Doxil transport to the tumor cells. Step 1: circulation of the doxorubicin-containing liposomes in the blood circulation with a half-life of approximately 55 hours (for humans) after injection without releasing the drug. Step 2: extravasation of ∼85 nm liposomal nanovehicles into the tissue compartment through the leaky tumor vasculature. Step 3: release of the free doxorubicin from the liposome, believed to be due to the physical and chemical breakdown of the liposomal membrane in the intestinal fluid because of the low pH, the presence of oxidizing agents and enzymes, or via the uptake by macrophages. Step 4: penetration of the free drug into the tumor cells, binding with the nucleic acid, followed by killing of the tumor cells. | ||
There has also been scientific focus on investigating and designing polymeric nanocarriers as a new area of nanomedicines and as efficient drug delivery vehicles. BIND-014 is a cancer drug formulation consisting of 100 nm polymeric nanospheres loaded with dacetaxel (a drug used to treat solid tumors). Like Doxil, this drug-carrying polymeric nanovehicle also relies on its size to leave the tumor vasculature.84,86 In BIND-014, the drug-carrying polymeric core has been engineered to control the release of the drug and the outer layer is composed of PEG and specific biomarkers to allow the nanocarrier to evade the body's immune system and at the same time to make it highly specific to the tumor cells. ABI-007 is another novel bioformulation that incorporates albumin particle technology and provides a novel treatment for breast cancer. ABI-007, also known as Abraxane, consists of 130 nm biologically interactive albumin-bound pacitaxel particles administered as a colloidal suspension.12 These nanosized drug carriers allow the safer infusion of significantly higher doses of pacitaxel than standard pacitaxel treatment, with shorter infusion schedules (30 min versus 3 h) and no premedication.87 In addition, as a result of their small size, the nanostructured ABI-007 colloidal particles are reportedly able to penetrate and reach deeper regions of solid tumors.
All these chemotherapeutic nanomedicines (Doxil, ∼85 nm; BIND-014, 100 nm; and ABI-007, 130 nm) with sizes of about 100 nm are reported to have significant antitumor activity, but only in highly vascularized tumors such as Kaposi's sarcoma and breast cancer. This is because, although a DDS size of about 100 nm is sufficient to seep out from the bloodstream into the blood vessels of the tumor and to treat permeable tumors, it is too large to penetrate deep into abnormal tissues of hypovascular and solid, impermeable pancreatic tumors. Cabral et al.88 investigated and compared the accumulation and effectiveness of drug-loaded polymer micelles with diameters of 30, 50, 70, and 100 nm in both highly and poorly permeable tumors. These studies showed that all the nanosized polymer micelles (30–100 nm) are equally competent and able to penetrate the highly permeable tumors. However, for poorly permeable tumors, only the smallest micelles 30 nm in size have succeeded in reaching deep inside poorly permeable pancreatic tissue to achieve the desired antitumor effect. Nishiyama et al.89 also attempted to target highly hypopermeable Lewis lung cancinoma cells using small (28 nm) cisplatin-incorporated polymeric micelles. Cisplatin is a platinum-based drug that has most commonly been used in pancreatic chemotherapy. These micelles have been found to show remarkably prolonged blood circulation with effective accumulation in solid tumors. Interestingly, in spite of their small size, these nanocarriers show reduced detrimental accumulation in vital organs compared with the free drug, an indication of their promising specificity and efficiency as drug nanocarriers. Nanocarrier, a company owned by Kazunori Kataoka, a materials scientist from the University of Tokyo (Japan), developed a 30 nm polymer DDS to transport cisplatin.90 The administration of free cisplatin in the body usually causes severe renal toxicity and requires the patient to drink large amounts of water during treatment. However, as a result of its small carrier size, this newly designed polymeric formulation allows the drug to accumulate more in the region of the pancreatic tumor instead of in the kidneys and provides an alternative to this excruciating treatment with enhanced survival time.
In conclusion, nanomedicines are now established nanocarriers for delivering chemotherapeutic drugs to regions of malignant tissue. The upper size range has been optimized to 300 nm, which is large enough to leave the leaky blood vessels and punctured lymphatic drainage at the tumor tissue site. These nanocarriers can be easily internalized by the MPS cells and therefore other body tissues are protected from the dose-dependent toxic effects of these drugs. The larger nanocarriers appear to stay near the tumor vasculature instead of diffusing throughout the tumor matrix. To enhance the bioavailability and accumulation of drugs in the tumor cells, nanovehicles of 50–150 nm have been proved to be effective. Nanomedicines in this size range, e.g. Doxil, BIND-014, and ABI-007 (Table 5), have already been successful in clinical settings and remarkable results have been obtained. A few other nanocarriers, such as NK105 and CALAA-01, are also currently in clinical trials. These nanomedicines are undoubtedly established as leaders in cancer therapeutics, but their efficacy is limited to treating permeable tumors, and they are still large enough to penetrate deep into the abnormal tissue foliage of hypovascular and solid impermeable tumors such as pancreatic tumors. To reach the deep vasculature of these hypopermeable tumors, it is necessary to reduce the size of the nanocarriers further to <50 nm. Daunoxome (45 nm) has already been approved by the FDA to target AIDS-related Kaposi's sarcoma and many such smaller sized drugs are in advanced clinical trials. Thus to capitalize on the EPR effect and to efficiently escape physiological barriers, many studies advocate an optimum nanoparticle size range of approximately 10–250 nm.91 Table 5 shows different FDA-approved nanomedicines (including their size and formulation) and other nanoscale drug carriers under clinical trial for cancer treatment.
| Nanomedicine | Drug formulation | Size (nm) | Present status | Description |
|---|---|---|---|---|
| Doxil | PEGylated doxorubicin-containing liposomes | ∼85 | First FDA-approved nanomedicine | Targets ovarian cancer, AIDS-related Kaposi's sarcoma and multiple myeloma with bortezomib |
| Myocet | Doxorubicin-containing liposomes | 150–250 | Approved in Europe and Canada for treatment of metastatic breast cancer in combination with cyclophosphamide, but is not yet approved by the FDA for use in the USA | Targets metastatic breast cancer. Taken up by mononuclear phagocytic cells due to comparatively large size |
| Daunoxome92 | Daunorubicin-containing liposomes | ∼45 | FDA-approved daunoxome as first-line treatment for Kaposi's sarcoma, but still under clinical trial for breast cancer treatment | Targets AIDS-related Kaposi's sarcoma and other blood cancers |
| BIND-014 | PEGylated dacetaxel containing polymeric spheres | 100 | In Phase I clinical trials | Targets solid or metastatic prostate cancer cells by binding to prostate-specific membrane antigen |
| ABI-007 | Albumin-bound pacitaxel particle | 130 | In Phase III clinical trials; recently approved by FDA | For patients with pre-treated metastatic breast cancer |
| NK10593 | Polymeric nanocarrier containing paclitaxel | 85 | Under Phase II clinical trials for stomach cancer and Phase III clinical trials for breast cancer | Progression-free survival in patients with metastatic or recurrent breast cancer |
| Nanoplatin (NC-6004) | PEGylated polymer nanocarrier containing cisplatin | 30 | Under Phase I/II clinical trials | For advanced or metastatic pancreatic cancer aiming for reduced kidney toxicity |
| Oxaliplatin94(NC4016) | PEGylated polymer micelles containing DACH-Pt | ∼30 | Under Phase I clinical trials | Platinum-based chemotherapy drug used in the treatment of colorectal cancer |
| CALAA-0195 | Cyclodextrin-based polymeric nanocarrier containing gene-silencing RNA | >100 | Under Phase I clinical trials | Holds RNA that silences a gene in solid tumors needed for DNA synthesis and replication |
| CRLX10196 | pH-sensitive cyclodextrin-based polymeric nanocarrier releases camptothecin in acidic environment of cancer cells | 25–50 | Under Phase II clinical trials | Separate studies testing CRLX101 in advanced non-small cell lung cancer and in ovarian cancer |
3.2 Effect of size of drug delivery vehicles on lung-targeted intravenous administration
Lung-targeted DDSs (LT-DDSs) are DDSs that can deliver the required medicine effectively to the lung to increase the drug concentration within a target zone and reduce the distribution of the drug to other organs or tissues. This aims to increase the efficacy of the drug, to minimize drug-originated systemic toxic effects, and to improve patient compliance and convenience. It is thus an ideal strategy for the treatment of lung disease. LT-DDSs mainly consist of dry powder formulations, nebulized suspensions, or inhaled solutions administered via the pulmonary route and liposomes, nanoparticles, or microparticles administered intravenously.97 Lung targeting via the pulmonary route in the context of the size of the DDS has been discussed in detail earlier. We provide here a brief overview of the effect of the size of the drug carrier on lung-targeted drug delivery via intravenous administration. Pulmonary administration has attracted much scientific attention for the delivery of drugs, but this has limitations. The main disadvantage is that most of the drugs need to be administered at least three to four times a day in aerosol form due to the short duration of the resultant clinical effects; in addition, the rapid absorption of some drugs, such as bronchodilators and corticosteroids, from the lung epithelium results in undesirable side-effects. Pulmonary delivery is not yet capable of delivering drugs to the location of disease in the lungs via inhalation due to the blocking of airways by inflammation or mucus plugs, which leads to more deposition in the conducting airways rather than in the peripheral airways. Therefore recent interest has focused on designing LT-DDSs that can be administrated via the intravenous route.Polymeric microparticles, including microspheres and microcapsules, are among the most studied drug carriers for lung-targeted drug delivery by the intravenous route. As reported previously, particles in the size range 7–28 μm are more likely to accumulate or deposit in the lungs by mechanical filtration through the capillary bed of the lung after intravenous administration.98,99 Hao et al.100 prepared and demonstrated the efficiency of ceftiofur-loaded gelatin microspheres with a mean diameter of 21.26 μm as a LT-DDS. Organ distribution pattern studies have shown a higher accumulation of the drug in the lungs than in any other organ (e.g. liver, spleen, stomach) with a lung-targeting efficiency (Te) enhancement of a factor of 300. With the objective of designing lung-targeted drug carriers, Yang et al.101 prepared erythromycin-loaded gelatin microspheres with a mean particle size of 15.62 μm. Drug distribution studies again showed significantly enhanced accumulation of the microparticles in the lungs over other tissues such as the liver, kidney, heart, spleen, and plasma. Tang et al.102 reported the chemical preparation of enrofloxacin-loaded lung-targeting microspheres by emulsification with gelatin and liquid paraffin. The microspheres were observed to have a mean diameter of 11.7 μm with 92.8% in the range 7–30 μm. After a single dose injection into dogs, the half-life of the distribution phase reduced by 77.78%, the half-life of the elimination phase lengthened from 5.15 to 33.86 h, and the clearance of the drug concentration in the lung reduced from 0.603 to 0.267 l h−1 kg−1 compared with the conventional drug. In addition, the relative intake rate (Re) and Te of the lung were 2.48 and 4.27, respectively, much greater than that of other tissues.
In another study, Sree et al.103 developed lung-targeting albumin-loaded ofloxacin microspheres (ALOME) composed of ofloxacin and albumin prepared by a water-in-oil emulsion method. The ALOME drug carriers, with an average particle size of 11.32 μm, showed the maximum drug concentration in the lung, i.e. 1048 μg g−1 at 10 min, significantly more than that in any other tissues and blood.33
With the aim of designing pactitaxel-based novel drug delivery vehicles, Yan and Pei104 prepared sustained release microspheres of paclitaxel using poly(1actic-co-glycolic acid) (PLGA) as the biodegradable material using emulsion evaporation technologies. The mean diameter of the microspheres was 9.65 mm, with over 87.2% of the microspheres ranging from 5 to 15 μm. They determined the drug concentration in the lung, heart, liver, spleen, and kidney of mice at 0.25, 1, 24, and 72 h after the intravenous administration of paclitaxel microspheres and with the injectable formulation as a control. The results showed that, compared with the injective solution, the drug concentration in the lung increased by as much as about 30 times in the case of the microspheres. Furthermore, the inhibition of growth rate in subcutaneously implanted tumors with the paclitaxel microspheres and injection were 83.1 and 62.8%, respectively. These results suggest that the microsphere carriers could target the delivery of paclitaxel to the lung and its increase antitumor efficiency.104
Using one of the most potent anticancer drugs, cisplatin, Huo et al.98 fabricated lung-targeting drug delivery PLGA microspheres with a diameter of 12.8 mm, with 98% of the microspheres in the range 5–30 mm. The drug distribution results with the cisplatin-loaded PLGA microsphere formulation showed a notably higher concentration of the drug in the lung (212 mg g−1, 15 min) than in other tissues and blood, whereas the drug concentration in the lung was only 1.37 mg g−1 15 min after the administration of a cisplatin injection in rabbits. Ying et al.105 prepared PLGA microcapsules containing carboplatin (a platinum-based antitumor drug) with a mean diameter of 14.25 ± 3.52 μm and studied their tissue distribution in mice after administration of a single intravenous dose. The results showed that the drug concentration of carboplatin microcapsules in the lungs increased by a factor of 1.76 compared with carboplatin solution as the controls. Lu et al.99 developed carboplatin gelatin microspheres with an average particle size of 13.20 μm, with 98% of the microspheres in the range 5.0–28.6 μm. The results showed that the Te of the lung increased 9.4 times compared with the spleen and 90.5 times compared with the liver for carboplatin microspheres. The results of pharmacodynamic study based on the mouse model of S-180 lung cancer suggested that the microcapsulated carrier improved the antitumor effect of carboplatin.
Guo et al.106 investigated the feasibility of the targeted delivery of protionamide (an anti-tuberculosis drug) to the lung through the intravenous administration of PLGA microspheres with a mean diameter of 9.86 ± 1.38 μm, with over 81.53% of the microspheres ranging from 7 to 15 μm. Compared with the aqueous formulation, the drug concentration in the lung of mice for the microsphere group increased by more than a factor of two at 6 h and was maintained for a long period. Wang et al.107 introduced docetaxel-loaded, glutaraldehyde cross-linked microspheres with a uniform size of 9.6 ± 0.8 μm as an LT-DDS. The microspheres released the maximum amount of the drug in the target tissue (the lungs). Table 6 shows different microparticles designed for lung-targeted drug delivery via intravenous administration.
| MP type | Active ingredient | Particle size (μm) | Lung-targeting effect | Animal model used | Reference |
|---|---|---|---|---|---|
| a MP: microparticles, MS: microspheres, Re: the relative intake rate, Te: targeting efficiency, Ce: ratio of drug peak concentration in lungs, PLGA:PLGA:poly(lactic-co-glycolic acid). | |||||
| Albumin | Ofloxacin | 11.32 | Concentration of drug in lung at 10 min: MS formulation 1048 μg g−1, control formulation 432 μg g−1 | Mouse | 103 |
| Gelatin | Enrofloxacin | 11.7 | Re 2.48, Te 4.27, Ce 4.27 | Dog | 102 |
| PLGA | Paclitaxel | 9.65 | Drug concentration showed 30 times increase in the lungs using MP formulation compared with injective drug solution in 15 min | Mouse | 104 |
| PLGA | Protionamide | 9.86 ± 1.38 | Drug concentration shows one-fold enhancement in the lungs using MP formulation compared with aqueous formulation at 6 h and maintained for long duration | Mouse | 106 |
| PLGA | Cisplatin | 12.8 | Concentration of drug in lung at 15 min: MS formulation 212 μg g−1, control formulation 1.37 μg g−1 | Rabbit | 98 |
| PLGA | Carboplatin | 14.25 ± 3.52 | Compared with control formulation Re 3.41, Te 2.82–5.58, Ce 1.76 | Mice | 105 |
| Gelatin | Ceftiofur | 21.26 | Te of lung increased by factor of 300 compared with blood and stomach, and 27.5 and 15.95 for liver and spleen | Mice | 100 |
| Gelatin | Erythromycin | 15.62 | Drug targeting index of MS formulation is 6.65 compared with erythromycin solution | Rabbit | 100 |
| Gelatin | Carboplatin | 13.20 | Ce 4.1, Te increased 9.4 times compared with spleen and 90.5 times compared with liver | Mice | 99 |
| Chitosan | Docetaxel | 9.6 ± 0.8 | MS are found to release the drug to a maximum extent in the target tissue | Mice | 107 |
In addition to microparticles, liposomes and nanoparticles have also been explored for their applications in intravenous mediated lung-targeted drug delivery. Many studies have shown that liposomes mostly accumulate in the organs of the reticuloendothelial system, such as the liver, spleen, and lung within the first 15–30 min after the intravenous administration of the liposomal formulation.108 In general, liposomes with a particle size >5 μm could be trapped passively by the vascular network of the lung to obtain a lung-targeting effect.46,109 With these size considerations, Cheng et al.110 developed dipyridamole (DIP) liposomes with a mean diameter of 4.434 ± 0.252 μm in a particle size range of 1.103 ± 0.080 and determined the drug (pyrimidopyrimidine) concentration in the lung, heart, liver, spleen, and kidney of mice at different time intervals after intravenous administration. The results showed that the relative intake rate for the lung was 2.23 in the case of DIP liposomes, which indicated that the exposure of DIP to the lung was significantly increased by a liposome carrier. The Te of the lung increased by a factor of 12.07 compared with plasma, 1.98 compared with the spleen, and 1.49 compared with the liver for DIP liposomes. Zhang et al.109 developed levofloxacin-loaded liposomes composed of soybean phosphatides, cholesterol, and levofloxacin by the ammonium sulfate gradient method. The results showed that the mean particle size and zeta potential of levofloxacin liposomes were 7.424 ± 0.689 μm and 13.11 ± 1.08 mV, respectively. They studied the tissue distribution of levofloxacin in rabbits after intravenous administration. The results suggested that the value of Re for the lung was 7.02, which was higher than for other tissues with the levofloxacin liposomes. Jiang et al.111 explored multivesicular liposomes loaded with gedopentetate dimeglumine, also known as DepoFoam, for its lung-targeted drug delivery characteristics. The biodistribution results obtained for this liposomal DepoFoam formulation (average diameter ∼18 μm) indicated that the concentration of the drug in the lung 30 min after intravenous administration was 325.17 ± 74.52 mg g−1 compared with 6.69 ± 1.82 mg g−1 for the conventional drug. Thus it was concluded that the liposomal formulation possesses a good lung-targeted effect.
It has been found that, in addition to the size of the liposomes, their disposition and localization is also affected by the surface charge and lipid composition.112,113 Several workers have reported that liposomes bearing a negative surface charge could accumulate in the lungs to a greater extent than the free drug or neutral or positively charged liposomes of similar size.114,115 In contrast with this, Jonah et al.116 found the highest uptake of EDTA drug by the lungs from positively charged liposomes after a single intravenous administration of a liposomal preparation. Wang et al.117 developed azithromycin liposomes with a mean particle size of 6.582 μm with a zeta potential of 19.5 mV. The azithromycin concentration in the heart, liver, spleen, lung, and kidney of mice after the intravenous administration of a liposomal and injectable formulation was then measured. The results showed that after intravenous injection into mice, the AUC in the lung increased 7.4 fold in the case of azithromycin liposomes compared with its solution. It is therefore thought that large particles with a positive charge could deliver the drug to the lung more easily than other particles. Zhao and coworkers118,119 used a combination of solid dispersion and effervescent techniques to prepare docetaxel liposomes composed of docetaxel/Tween-80/Phosopholipon90H/cholesterol/citric acid at molar ratios of 0.18:0.09:3.78:3.78:91.17 with a diameter of 1011 ± 22 nm. The zeta potential and entrapment efficiency of the resulted liposomes were −23.7 ± 0.26 mV and 90.12 ± 0.36%, respectively. In general, for the passive targeting of liposomes to the lungs, the particle size should be >5 μm to be retained in the alveolar capillaries. Interestingly, when the evaluation of the lung-targeting effect of docetaxel liposomes in rabbits was studied, it was found that negatively charged docetaxel liposomes with a diameter of about 1 μm had a favorable lung-targeting effect; the Re and the Ce (ratio of peak concentration) of the lung were 28.91 and 74.28, respectively. Accepting that for passive targeting to the lungs the size of the DDS should be above ≥7 μm, there are few published reports on lung-targeted DDSs of nanoparticles via the intravenous route of administration except in cancer therapeutics.
Therefore, as particles in the size range 7–28 μm have a greater tendency to accumulate or deposit in the lungs by mechanical filtration after intravenous administration, particles ≥7 μm are the most suitable for lung-targeted intravenous drug delivery. However, this particle dimension can vary depending on the carrier surface properties (such as charge, hydrophobic/hydrophilic properties, or porosity).
4. Conclusions
This review paper has attempted to provide a comprehensive overview of the effect of the size of the drug delivery vehicle on its biodistribution, specific organ targeting, body clearance kinetic, and, most importantly, therapeutic efficiency. The key emphasis has been to compile the relevant published papers, particularly in the field of pulmonary and intravenous administration, focusing mainly on cancer therapeutics and lung-targeted drug delivery. For pulmonary drug administration, drug carriers of 1–5 μm are confirmed to be the optimum size. Smaller delivery vectors have cytotoxic effects and most (80%) are removed by exhalation without being deposited or conveying any therapeutic effect. In contrast, larger particles (>5 μm) are cleared out by a strong mucociliary clearance mechanism. For intravenous drug delivery, the size of the drug delivery vehicle depends on the organs targeted, their endothelial fenestration size, and on internalization. For cancer therapeutics, nanomedicines, particularly drug-entrapped liposomes, polymeric nanoparticles, and dendrites, have been studied by many research groups. The effective size range for the drug delivery carriers targeting tumor cells are in the range 10–250 nm depending on the type of the targeted tumor cells (hypopermeable/hyperpermeable or hypovascularized/hypervascularized), the tumor location, and the composition and surface characteristics of the carrier material. Microparticles ≥7 μm have proved to be the best vehicles for lung-targeted intravenous administration. This review attempts to highlight a very important physical parameter related to drug carriers, which has a pivotal role to play in drug therapeutic, i.e. the size of the drug delivery carrier.Acknowledgements
We acknowledge the financial support received from the Singapore Ministry of Health's National Medical Research Council under its Cooperative Basic Research Grant (NMRC/CBRG/0048/2013) and TCR Grant R618/41/2008 (RWB), R1054/69/2013 (RL).References
- G. Tiwari, R. Tiwari, B. Sriwastawa, L. Bhati, S. Pandey, P. Pandey and S. K. Bannerjee, Int. J. Pharm. Invest., 2012, 2, 2–11 CrossRef PubMed.
- M. Koval, K. Preiter, C. Adles, P. D. Stahl and T. H. Steinberg, Exp. Cell Res., 1998, 242, 265–273 CrossRef CAS PubMed.
- L. Illum, S. S. Davis, C. G. Wilson, N. W. Thomas, M. Frier and J. G. Hardy, Int. J. Pharm., 1982, 12, 135–146 CrossRef CAS.
- S. Bamrungsap, Z. Zhao, T. Chen, L. Wang, C. Li, T. Fu and W. Tan, Nanomedicine, 2012, 7, 1253–1271 CrossRef CAS PubMed.
- V. Morigi, A. Tocchio, C. B. Pellegrini, J. H. Sakamoto, M. Arnone and E. Tasciotti, J. Drug Delivery, 2012, 2012, 1–7 CrossRef PubMed.
- J. A. Junghanns and R. H. Muller, Int. J. Nanomed., 2008, 3, 295–310 CAS.
- H. I. Chang and M. K. Yeh, Int. J. Nanomed., 2012, 7, 49–60 CAS.
- M. L. Immordino, F. Dosio and L. Cattel, Int. J. Nanomed., 2006, 1, 297–315 CrossRef CAS PubMed.
- T. M. Allen and P. R. Cullis, Science, 2004, 303, 1818–1822 CrossRef CAS PubMed.
- J. Khandare and T. Minko, Prog. Polym. Sci., 2006, 31, 359–397 CrossRef CAS PubMed.
- P. Kesharwani, K. Jain and N. K. Jain, Prog. Polym. Sci., 2014, 39, 268–307 CrossRef CAS PubMed.
- W. J. Gradishar, S. Tjulandin, N. Davidson, H. Shaw, N. Desai, P. Bhar, M. Hawkins and J. O. Shaughnessy, J. Clin. Oncol., 2005, 23, 7794–7803 CrossRef CAS PubMed.
- J. Alder-Moore, Bone Marrow Transplant., 1994, 14, S3–S7 Search PubMed.
- S. Venkataraman, J. L. Hedrick, Z. Y. Ong, C. Yang, P. L. Rachel Ee, P. T. Hammond and Y. Y. Yang, Adv. Drug Delivery Rev., 2011, 63, 1228–1246 CrossRef CAS PubMed.
- J. A. Champion, Y. K. Katare and S. Mitragotri, J. Controlled Release, 2007, 121, 3–9 CrossRef CAS PubMed.
- S. Honary and F. Zahir, Trop. J. Pharm. Res., 2013, 12, 255–264 Search PubMed.
- S. Honary and F. Zahir, Trop. J. Pharm. Res., 2013, 12, 265–273 Search PubMed.
- J. W. Yoo, N. Doshi and S. Mitragotri, Adv. Drug Delivery Rev., 2011, 63, 1247–1256 CrossRef CAS PubMed.
- R. A. Petros and J. M. DeSimone, Nat. Rev. Drug Discovery, 2010, 9, 615–627 CrossRef CAS PubMed.
- S. Nagayama, K. I. Ogawara, Y. Fukuoka, K. Higaki and T. Kimura, Int. J. Pharm., 2007, 342, 215–221 CrossRef CAS PubMed.
- D. C. Litzinger, A. M. J. Buiting, N. V. Rooijen and L. Huang, Biochim. Biophys. Acta, Biomembr., 1994, 1190, 99–107 CrossRef CAS.
- Y. Matsumura and H. Maeda, Cancer Res., 1986, 46, 6387–6392 CAS.
- W. Jiang, B. Y. Kim, J. T. Rutka and W. C. Chan, Nat. Nanotechnol., 2008, 3, 145–150 CrossRef CAS PubMed.
- J. S. Patton, Adv. Drug Delivery Rev., 1996, 19, 3–36 CrossRef CAS.
- I. Sermet-Gaudelus, Y. L. Cocquic, A. Ferroni, M. Clairicia, J. Barthe, J. P. Delaunay, V. Brousse and G. Lenoir, Pediatr. Drugs, 2002, 4, 455–467 CrossRef PubMed.
- J. S. Patton, C. S. Fishburn and J. G. Weers, Proc. Am. Thorac. Soc., 2004, 1, 338–344 CrossRef CAS PubMed.
- J. S. Patton and P. R. Byron, Nat. Rev. Drug Discovery, 2007, 6, 67–74 CrossRef CAS PubMed.
- J. H. Hamman, G. M. Enslin and A. F. Kotze, BioDrugs, 2005, 19, 165–177 CrossRef CAS PubMed.
- F. Chellat, Y. Merhi, A. Moreau and L. Yahia, Biomaterials, 2005, 26, 7260–7275 CrossRef CAS PubMed.
- K. C. Stone, R. R. Mercer, P. Gehr, B. Stockstill and J. D. Crapo, Am. J. Respir. Cell Mol. Biol., 1992, 6, 235–243 CrossRef CAS PubMed.
- D. A. Groneberg, C. Witt, U. Wagner, K. F. Chung and A. Fischer, Respir. Med., 2003, 97, 382–387 CrossRef CAS PubMed.
- H. M. Courrier, N. Butz and T. F. Vandamme, Crit. Rev. Ther. Drug Carrier Syst., 2002, 19, 425–498 CrossRef CAS.
- J. S. Patton, J. D. Brain, L. A. Davies, J. Fiegel, M. Gumbleton, K. Jin Kim, M. Sakagami, R. Vanbever and C. Ehrhardt, J. Aerosol Med. Pulm. Drug Delivery, 2010, 23, S71–S87 CAS.
- T. B. Martonen and I. M. Katz, Pharm. Res., 1993, 10, 871–878 CrossRef CAS.
- P. L. Ariyananda, J. E. Agnew and S. W. Clarke, Postgrad. Med. J., 1996, 72, 151–156 CrossRef CAS.
- W. C. Hinds, Aerosol technology: properties, behavior, and measurement of airborne particles, Wiley Publications, 2 edn, 1999 Search PubMed.
- P. R. Byron, M. Hindle, C. F. Lange, P. W. Longest, D. McRobbie, M. J. Oldham, B. Olsson, C. G. Thiel, H. Wachtel and W. H. Finlay, J. Aerosol Med. Pulm. Drug Delivery, 2010, 23, S59–S69 CrossRef CAS PubMed.
- S. Shi, E. S. Ashley, B. D. Alexander and A. J. Hickey, AAPS PharmSciTech, 2009, 10, 129–137 CrossRef CAS PubMed.
- J. Heyder and G. Rudolf, J. Aerosol Sci., 1984, 15, 697–707 CrossRef.
- W. Stahlhofen, R. Koebrich, G. Rudolf and G. Scheuch, J. Aerosol Med., 1990, 21, S407–S410 CrossRef.
- N. Y. Yoo, Y. S. Youn, N. M. Oh, K. T. Oh, D. K. Lee, K. H. Cha, Y. T. Oh and E. S. Lee, Colloids Surf., B, 2011, 88, 419–424 CrossRef CAS PubMed.
- K. Hirota, T. Kawamoto, T. Nakajima, K. Makino and H. Terada, Colloids Surf., B, 2013, 105, 92–97 CrossRef CAS PubMed.
- K. Hirota, T. Hasegawa, H. Hinata, F. Ito, H. Inagawa, C. Kochi, G. Soma, K. Makino and H. Terada, J. Controlled Release, 2007, 119, 69 CrossRef CAS PubMed.
- J. H. Widdicombe and J. G. Widdicombe, Respir. Physiol., 1995, 99, 3–12 CrossRef CAS.
- D. Stuart, R. Lobenberg, T. Ku, S. Azarmi, L. Ely, W. Roa and E. J. Prenner, J. Biomed. Nanotechnol., 2006, 2, 245–252 CrossRef CAS PubMed.
- P. Borm, F. C. Klaessig, T. D. Landry, B. Moudgil, J. Pauluhn, K. Thomas, R. Trottier and S. Wood, Toxicol. Sci., 2006a, 90, 23–32 Search PubMed.
- J. S. Patton and P. R. Byron, Nat. Rev. Drug Discovery, 2007, 6, 67–74 CrossRef CAS PubMed.
- A. J. Bitonti, J. A. Dumont, S. C. Low, R. T. Peters, K. E. Kropp, V. J. Palombella, J. M. Stattel, Y. Lu, C. A. Tan, J. J. Song, A. M. Garcia, N. E. Simister, G. M. Spiekermann, W. I. Lencer and R. S. Blumberg, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 9763–9768 CrossRef CAS PubMed.
- M. Arredouani, Z. Yang, Y. Ning, G. Qin, R. Soininen, K. Tryggvason and L. Kobzik, J. Exp. Med., 2004, 200, 267–272 CrossRef CAS PubMed.
- J. Heyder, J. Gebhart, G. Rudolf, C. F. Schiller and W. Stahlhofen, J. Aerosol Sci., 1986, 17, 811–825 CrossRef.
- Y. Sibille and H. Y. Reynolds, Am. Rev. Respir. Dis., 1990, 141, 471–501 CrossRef CAS PubMed.
- S. Chono, T. Tanino, T. Seki and K. Morimoto, J. Drug Targeting, 2006, 14, 557–566 CrossRef CAS PubMed.
- J. M. Lauweryns and J. H. Baert, Am. Rev. Respir. Dis., 1977, 115, 625–683 CAS.
- J. Rejman, V. Oberle, I. S. Zuhorn and D. Hoekstra, Biochem. J., 2004, 377, 159–169 CrossRef CAS PubMed.
- M. Gumbleton, Adv. Drug Delivery Rev., 2001, 49, 281–300 CrossRef CAS.
- J. S. Patton, Adv. Drug Delivery Rev., 1996, 19, 3–36 CrossRef CAS.
- A. Nemmar, H. Vanbilloen, M. F. Hoylaerts, P. H. Hoet, A. Verbruggen and B. Nemery, Am. J. Respir. Crit. Care Med., 2001, 164, 1665–1668 CrossRef CAS PubMed.
- G. Oberdorster, Inhalation Toxicol., 2002, 14, 29–56 CrossRef CAS PubMed.
- W. G. Kreyling, M. Semmler, F. Erbe, P. Mayer, S. Takenaka, H. Schulz, G. Oberdorster and A. Ziesenis, J. Toxicol. Environ. Health, Part A, 2002, 65, 1513–1530 CrossRef CAS PubMed.
- G. Oberdorster, J. Ferin and B. E. Lehnert, Environ. Health Perspect., 1994, 102, 173–179 CrossRef.
- S. M. Moghimi and A. C. Hunter, Crit. Rev. Ther. Drug Carrier Syst., 2001, 18, 527–550 CrossRef CAS.
- P. Caliceti and F. M. Veronese, Adv. Drug Delivery Rev., 2003, 55, 1261–1277 CrossRef CAS.
- Y. Takakura, R. I. Mahato and M. Hashida, Adv. Drug Delivery Rev., 1998, 34, 93–108 CrossRef CAS.
- R. L. Conhaim, A. Eaton, N. C. Staub and T. D. Heath, J. Appl. Physiol., 1988, 64, 1134–1142 CAS.
- S. M. Moghimi, Adv. Drug Delivery Rev., 1995, 17, 61–73 CrossRef CAS.
- L. W. Seymour, Crit. Rev. Ther. Drug Carrier Syst., 1992, 9, 135–187 CAS.
- L. Cucullo, M. S. McAllister, K. Kight, L. Krizanac-Bengez, M. Marroni, M. R. Mayberg, K. A. Stanness and D. Janigro, Brain Res., 2002, 951, 243–254 CrossRef CAS.
- R. K. Jain, Sci. Am., 1994, 271, 58–65 CrossRef CAS PubMed.
- K. E. Arfors, G. Rutili and E. Svensjo, Acta Physiol. Scand., Suppl., 1979, 463, 93–103 CAS.
- R. L. Juliano and D. Stamp, Biochem. Biophys. Res. Commun., 1975, 63, 651–658 CrossRef CAS.
- S. Muro, C. Garnacho, J. A. Champion, J. Leferovich, C. Gajewski, E. H. Schuchman, S. Mitragotri and V. R. Muzykantov, Mol. Therapy, 2008, 16, 1450–1458 CrossRef CAS PubMed.
- M. Kanke, G. H. Simmons, D. L. Weiss, B. A. Bivins and P. P. Deluca, J. Pharm. Sci., 1980, 69, 755–762 CrossRef CAS.
- H. S. Choi, W. Liu, P. Misra, E. Tanaka, J. P. Zimmer, B. I. Ipe, M. G. Bawendi and J. V. Frangioni, Nat. Biotechnol., 2007, 25, 1165–1170 CrossRef CAS PubMed.
- S. M. Moghimi, A. C. Hunter and J. C. Murray, Pharmacol. Rev., 2001, 53, 283–318 CAS.
- R. Minchin, Nat. Nanotechnol., 2008, 3, 12 CrossRef CAS PubMed.
- http://www.drugs.com/news/global-market-cancer-therapies-slated-growth-through-2013-8407.html.
- J. A. Sparano and E. P. Winer, Semin. Oncol., 2001, 28, 32–40 CrossRef CAS PubMed.
- G. N. Hortobagyi, Drugs, 1997, 54, 1–7 CrossRef CAS PubMed.
- T. M. Allen and F. J. Martin, Semin. Oncol., 2004, 31, 5–15 CrossRef CAS PubMed.
- P. R. Kulkarni, J. D. Yadav and K. A. Vaidya, Curr. Pharm. Des., 2004, 10, 2981–2989 CrossRef.
- G. Batist, G. Ramakrishnan, C. S. Rao, A. Chandrasekharan, J. Gutheil, T. Guthrie, P. Shah, A. Khojasteh, M. K. Nair, K. Hoelzer, K. Tkaczuk, Y. C. Park and L. W. Lee, J. Clin. Oncol., 2001, 19, 1444–1454 CAS.
- F. Martin, in Medical applications of liposomes, ed. D. D. Lasic and D. Papahadjopoulos, Elsevier, New York, 1998, p. 638 Search PubMed.
- F. Yuan, M. Dellian, D. Fukumura, M. Leunig, D. A. Berk, V. P. Torchilin and R. K. Jain, Cancer Res., 1995, 55, 3752–3756 CAS.
- K. Bourzac, Nature, 2012, 491, S58–S60 CrossRef.
- P. S. Gill, J. Wernz, D. T. Scadden, P. Cohen, G. M. Mukwaya, J. H. Von Roenn, M. Jacobs, S. Kempin, I. Silverberg, G. Gonzales, M. U. Rarick, A. M. Myers, F. Shepherd, C. Sawka, M. C. Pike and M. E. Ross, J. Clin. Oncol., 1996, 14, 2353–2364 CAS.
- J. Hrkach, D. V. Hoff, M. M. Ali, E. Andrianova, J. Auer1, T. Campbell, D. D. Witt, M. Figa, M. Figueiredo, A. Horhota, S. Low, K. McDonnell, E. Peeke, B. Retnarajan, A. Sabnis, E. Schnipper, J. J. Song, Y. H. Song, J. Summa, D. Tompsett, G. Troiano, T. V. G. Hoven, J. Wright, P. LoRusso, P. W. Kantoff, N. H. Bander, C. Sweeney, O. C. Farokhzad, R. Langer and S. Zale, Sci. Transl. Med., 2012, 4, 128ra39 Search PubMed.
- N. Desai, V. Trieu, Z. Yao, L. Louie, S. Ci, A. Yang, C. Tao, T. De, B. Beals, D. Dykes, P. Noker, R. Yao, E. Labao, M. Hawkins and P. Soon-Shiong, Clin. Cancer Res., 2006, 12, 1317–1324 CrossRef CAS PubMed.
- H. Cabral, Y. Matsumoto, K. Mizuno, Q. Chen, M. Murakami, M. Kimura, Y. Terada, M. R. Kano, K. Miyazono, M. Uesaka, N. Nishiyama and K. Kataoka, Nat. Nanotechnol., 2011, 6, 815–823 CrossRef CAS PubMed.
- N. Nishiyama, S. Okazaki, H. Cabral, M. Miyamoto, Y. Kato, Y. Sugiyama, K. Nishio, Y. Matsumura and K. Kataoka, Cancer Res., 2003, 63, 8977–8983 CAS.
- H. Uchino, Y. Matsumura, T. Negishi, F. Koizumi, T. Hayashi, T. Honda, N. Nishiyama, K. Kataoka, S. Naito and T. Kakizoe, Br. J. Cancer, 2005, 93, 678–687 CrossRef CAS PubMed.
- F. Alexis, E. Pridgen, L. K. Molnar and O. C. Farokhzad, Mol. Biopharm., 2008, 5, 505–515 CAS.
- P. Paolo, Leukemia, 2002, 16, 1880–1881 CrossRef PubMed.
- T. Hamaguchi, Y. Matsumura, M. Suzuki, K. Shimizu, R. Goda, I. Nakamura, I. Nakatomi, M. Yokoyama, K. Kataoka and T. Kakizoe, Br. J. Cancer, 2005, 92, 1240–1246 CrossRef CAS PubMed.
- http://www.nanocarrier.co.jp/en/research/pipeline/03.html.
- A. C. Eifler and C. S. Thaxton, Methods Mol. Biol., 2011, 726, 325–338 CAS.
- T. Schluep, J. Hwang, J. Cheng, J. D. Heidel, D. W. Bartlett, B. Hollister and M. E. Davis, Clin. Cancer Res., 2006, 12, 1601–1606 CrossRef PubMed.
- Y. Wei and L. Zhao, Pharm. Dev. Technol., 2014, 19, 129–136 CrossRef CAS PubMed.
- D. J. Huo, S. H. Deng, L. B. Li and J. Ji, Int. J. Pharm., 2005, 289, 63–67 CrossRef CAS PubMed.
- B. Lu, J. Q. Zhang and H. Yang, Int. J. Pharm., 2003, 265, 1–11 CrossRef CAS.
- Z. Hao, B. Qu, Y. Wang, S. Tang, G. Wang, M. Qiu, R. Zhang, Y. Liu and X. Xiao, Drug Dev. Ind. Pharm., 2011, 37, 1422–1428 CrossRef CAS PubMed.
- F. Yang, S. G. Wu and Y. F. Pan, Drug Dev. Ind. Pharm., 2009, 35, 639–645 CrossRef CAS PubMed.
- S. Tang, Y. Zhou, R. Li, Q. Chen and X. Xiao, J. Vet. Pharmacol. Ther., 2007, 30, 443–450 CrossRef CAS PubMed.
- H. Sree, R. Chandramouli and R. Shobha, Int. J. Pharm., 2009, 380, 127–132 CrossRef PubMed.
- Z. Yan and Y. Y. Pei, Chin. J. Clin. Pharmacol., 2006, 15, 226–231 CAS.
- Y. Ying, S. W. Zhou, J. L. Tang, Y. Huang and Y. Xu, China Pharm., 2007, 18, 28–30 CAS.
- Z. Y. Guo, W. J. Zhao and Q. Zhang, Tuber. Thor. Tumor, 2011, 1, 19–24 Search PubMed.
- H. Wang, X. Yongdong and X. Zhou, Int. J. Mol. Sci., 2014, 15, 3519–3532 CrossRef PubMed.
- P. G. Waser, U. Muller, J. Kreuter, S. Berger, K. Munz, E. Kaiser and B. Pfluger, Int. J. Pharm., 1987, 39, 213–227 CrossRef CAS.
- X. Zhang, P. Sun, R. Bi, J. Wang, N. Zhang and G. Huang, J. Drug Target, 2009, 17, 399–407 CrossRef CAS PubMed.
- J. Cheng, N. Wen, F. Xiong, S. Chen and B. Zhu, J. Drug Target, 2006, 14, 717–724 CrossRef CAS PubMed.
- Q. J. Jiang, F. Geng and W. Zhang, Chinese J. Oncoradiology, 2009, 2, 25–29 Search PubMed.
- K. Hirano and C. A. Hunt, J. Pharm. Sci., 1985, 74, 915–921 CrossRef CAS.
- E. Sada, S. Kato, M. Terashima and H. Kawahara, J. Pharm. Sci., 1990, 79, 232–235 CrossRef CAS.
- R. M. Abra, C. A. Hunt and D. T. Lau, J. Pharm. Sci., 1984, 73, 203–206 CrossRef CAS.
- C. K. Kim, M. K. Lee, J. H. Han and B. J. Lee, Int. J. Pharm., 1994, 108, 21–29 CrossRef CAS.
- M. M. Jonah, E. A. Cerny and Y. E. Rahman, Biochim. Biophys. Acta, 1975, 401, 336–348 CrossRef CAS.
- J. S. Wang, J. B. Zhu and W. Shen, Acta Pharm. Sin. B, 2005, 40, 274–178 CAS.
- L. Zhao, Y. Ye, J. Li and Y. M. Wei, J. Pharm. Pharmacol., 2011, 63, 80–86 CrossRef CAS PubMed.
- L. Zhao, Y. M. Wei, X. D. Zhong, Y. Liang, X. M. Zhang, W. Li, B. B. Li, Y. Wang and Y. Yu, J. Pharm. Biomed. Anal., 2009, 49, 989–996 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |