
Polymethacrylic acid–facilitated nanofiber matrix loading Ag nanoparticles for SERS measurements†
Abstract
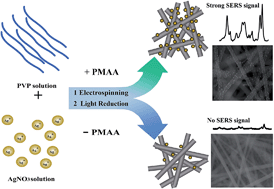
Nanofiber matrix loading Ag nanoparticles have been applied for SERS measurements with the limitation of poor reproducibility. By introducing polymethacrylic acid (PMAA) into the electrospun solutions in this contribution, fairly uniform PMAA/poly(N-vinylpyrrolidone) (PVP) ultrafine fibers containing silver nanoparticles (AgNPs) were successfully prepared via electrospinning by means of in situ photo reduction of silver ions. PMAA can significantly improve the absorbing amounts of silver ions in the polymer owing to its linear structure with abundant carboxyl groups, which makes the content and size of formed AgNPs in the polymer matrix to be easily controlled under different light sources (such as desk lamp, 365 nm UV lamp, and 254 nm UV lamp). With the electrospun AgNPs/PMAA/PVP fibrous membranes, malachite green (MG), a significant environmental organic pollutant known for its genotoxicity, was successfully detected with RSD values below 0.2% through SERS signals.
 Please wait while we load your content...
Please wait while we load your content...

