
Noncovalent assembly of carbon nanoparticles and aptamer for sensitive detection of ATP†
Jinhua Liu*ab,
Jing Yua,
Jianrong Chen*a and
Kaimin Shih*b
aCollege of Geography and Environmental Science, Zhejiang Normal University, Jinhua, 321004, People's Republic of China. E-mail: liujh@zjnu.cn; cjr@zjnu.cn; Fax: +86-579-82282269; Tel: +86-579-82282269
bDepartment of Civil Engineering, The University of Hong Kong, Pokfulam Road, Hong Kong, Hong Kong SAR, China. E-mail: kshih@hku.hk; Fax: +86-579-82282269; Tel: +86-579-82282269
First published on 18th August 2014
Abstract
Coupling carbon nanomaterials with biomolecular recognition events for sensor design has attracted great interest in the development of efficient bioanalytical tools. Here, based on competitive interaction of electrostatic repulsion and π–π stacking, noncovalent assembly of carboxylated carbon nanoparticles (cCNPs) with aptamer that allows sensitive and selective detection of ATP is reported. The sensor exhibits minimal background fluorescence, due to the extraordinarily high quenching efficiency of cCNPs with a spherical structure. Importantly, the quenched fluorescence is recovered with the addition of ATP within several minutes; the limit of detection is as low as 0.1 μM in the range of 0.1–300 μM, since only one end of the aptamer needs the modification, the present approach is simple and cost-effective. Furthermore, compared to the analog design based on the “pre-mixing” strategy, the assay of the “post-mixing” strategy increases by approximately 1.5 times in signal-to-background (S/B) and possesses a quicker response time (within two minutes). Depending on the spherical structure of the cCNPs and the rapid kinetic response, this assay can be expected to provide a new and ultrasensitive platform for the detection of various small molecules.
1. Introduction
Adenosine-5′-triphosphate (ATP) is a universal energy currency of living organisms, which plays a critical role in the regulation of cellular metabolism and biochemical pathways in cell physiology,1,2 such as in regulation of tumor growth, metastasis, and angiogenesis.3–5 In addition, it has had widespread use as an indicator for cell viability and cell injury in living organisms.6 Therefore, sensitive and selective determination of ATP is essential for biochemical study and clinical diagnosis.Aptamers are artificial oligonucleotides (DNA or RNA) that can bind to a wide variety of entities (metal ions, small organic molecules, proteins, and cells) with high selectivity, specificity, and affinity, equal to or often superior to those of antibodies.7 These binding features of aptamers are employed for the development of different sensor systems, for example, aptamer-based sensors (aptasensors) have been extensively used in the detection of cancer cells, drugs, and a variety of proteins.8 Up to now, the detection of ATP based on its aptamer is one of mostly used strategies because of the excellent specificity of aptamers toward ATP.9,10 Numerous aptasensors coupling carbon nanomaterials have been exploited by the binding-induced conformational changes to monitor the interaction with targets by fluorescence quenching,11,12 among them carbon nanotube and graphene oxide have also been successfully designed as a platform for application in ATP detection.13,14 Although these nanomaterials have been demonstrated to be chemically inert, they are hydrophobic with huge specific surface areas, so that the adsorption behavior of the DNA probe is mainly determined by strong hydrophobic and π–π stacking interactions.15,16 The high-energy barrier of the huge specific surface area of graphite precludes the possibility of obtaining acceptable endpoints,16,17 thus limiting their applications in rapid and real-time bioanalysis. Specifically, a promising application of carbon nanoparticles in fluorescent sensing technology is because of it being a good energy acceptor in energy transfer. It has been shown that carbon nanoparticle can function as both a “nano-scaffold” for oligonucleotides and a “nano-quencher” of dyes.18–20 Recent works have reported the noncovalent assembly of carbon nanoparticles and aptamer for detection of DNA, Hg2+, thrombin and DNA methylation.21 However, detection of small molecule using cCNPs as a platform of aptasensor has rarely been reported.
Herein, based on the electrostatic repulsion and π–π stacking interactions between carbon nanoparticles and oligonucleotides, we report a sensitive, selective and stable high-performance sensor for ATP detection. The sensing principle (Fig. 1) is in view of the “post-mixing” strategy: when P is hybridized with its target ATP to form a complex, the fluorescence mostly remains in the presence of cCNPs. It is quite different from the “pre-mixing” strategy: firstly, the aptamer–ATP (P) strand binds to the cCNPs surface strongly to form P–cCNPs complex, and then significant fluorescence is restored by the addition of ATP via time. In direct contrast, the cCNPs aptasensor of the “post-mixing” strategy can detect 0.1 μM ATP with good selectivity, and the S/B ratio is 1.5-fold more than that of the “pre-mixing” strategy. Importantly, noncovalent assembly of carbon nanoparticles and aptamer using the “post-mixing” strategy for ATP detection solves suffered key issues: firstly, the preparation method of cCNPs is simple; secondly, the prepared cCNPs are dispersed well in water without using the surfactant, although the surfactant can make the graphene oxide disperse well in solution; it will disturb the property of carbon nanomaterials.22 Finally, the S/B is very high (up to 7) because of low-background fluorescence. All these suggest the cCNPs aptasensor based on “post-mixing” strategy shows excellent promising in biomolecular detection.
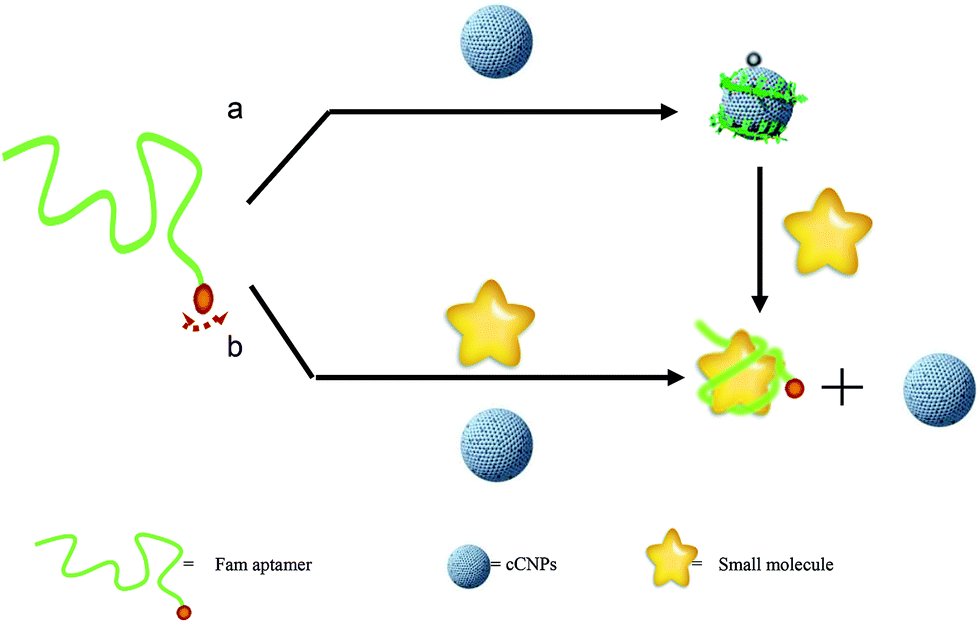 | ||
| Fig. 1 Schematic description of aptamer fluorescent sensing based on the cCNPs: (a) dye-tagged aptamer and cCNPs are mixed to form the cCNP–aptamer complex and the dye fluorescence are quenched, and then addition of ATP resulting in the restoration of the dye fluorescence. (b) Dye-tagged aptamer was hybridized with ATP to form a complex, and then addition of cCNPs the dye fluorescence mostly remains. | ||
2. Experimental section
Chemical and apparatus
All the DNA synthesis reagents were purchased from Glen Research. All oligonucleotides with different sequences were synthesized using an ABI3400 DNA/RNA synthesizer and dissolved in highly pure water (sterile Millipore water, 18.3 MΩ) as the stock solution. Fluorescence measurements and fluorescence anisotropy measurements were performed using a Photo Technology Intl (U.S.A.). The microstructures of the cCNPs were examined using a JEOL JSM-6700F scanning electron microscope (SEM) (Japan). A multimode AFM (SPI3800N-SPA400, Seiko Instruments, Japan) have a piezoscanner with a maximum scan range of 100 μm × 100 μm × 5 μm; XPS measurements were carried out on an Axis ultra imaging photoelectron spectrometer (Kratos Analytical Ltd., UK). ATP aptamer was synthesized by Shanghai Biotech (China) and labeled in the end with FAM dye. DNA sequence of aptamer–ATP was P: (5′-FAM-ACCTGGGGGAGTATTGCGGAGG AAGGT-3′). ATP, ADP, GTP and CTP were bought from Sigma (U.S.A.).The preparation of cCNPs
Pristine CNPs (pCNPs) were obtained from candle soot.23 To prepare oxidized cCNPs, 4.0 mg pristine of CNPs was mixed with 2.0 mL HNO3 (63%) and 2.0 mL DMF, and stirred for 24 h at 60 °C. After cooling down to room temperature, the upper 80% of the clear solution was removed by ultracentrifugation and the resulting dark powders were collected. The collected precipitates were cleaned with distilled water and subsequently centrifuged at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min more than three times. After being dried at 60 °C for 3 h, 2.4 mg oxidized cCNPs were obtained. We prepared the cCNPs stock solution by dissolving 2.0 mg of the oxidized cCNPs powders in 1.0 mL water, and the concentration of cCNPs solution of 2.0 mg mL−1 was received.
000 rpm for 10 min more than three times. After being dried at 60 °C for 3 h, 2.4 mg oxidized cCNPs were obtained. We prepared the cCNPs stock solution by dissolving 2.0 mg of the oxidized cCNPs powders in 1.0 mL water, and the concentration of cCNPs solution of 2.0 mg mL−1 was received.
Fluorescence measurements
The working solutions of the fluorescent oligonucleotides were obtained by diluting the stock solution to about 30 nM with the Tris–HCl buffer. To study the kinetics and time dependence of the fluorescence quenching of the fluorescent aptamer (P, 5′-FAM-ACCTGGGGGAGTATTG-CGGAGG AAGGT-3′) by cCNPs, cCNPs were first sonicated in doubly deionized water for 2 h to give a homogeneous black solution. After the pretreatment, an aliquot of the freshly made cCNPs suspension (less than 1%, v/v) was added to 500 μL of Tris–HCl buffer containing 30 nM of P, and the level of fluorescence emission intensity was then recorded with time. For target detection, different concentrations of ATP were first interacted with the aptamer for five minutes at room temperature and then recorded with the addition of an aliquot of the freshly made cCNPs suspension. We evaluated the response sensitivity in terms of the signal-to-background ratio (S/B), which was defined as S/B = (Fhybrid − Fbuffer)/(Fprobe − Fbuffer), where Fprobe, Fbuffer, and Fhybrid are the fluorescence intensities of the probes without target, plain buffer solution, and the probe–target hybrid, respectively.The definition of “pre-mixing” and “post-mixing” strategy
The “pre-mixing” strategy: dye-tagged aptamer and cCNPs are mixed to form the cCNPs–aptamer complex and the dye fluorescence are quenched, and then addition of ATP resulting in the restoration of the fluorescence of the dye. The “post-mixing” strategy: dye-tagged aptamer is hybridized with ATP to form a complex, and then addition of cCNPs the fluorescence of dye largely remains.3. Results and discussion
3.1. Characterization of the carbon nanoparticles
The cCNPs were prepared via oxidizing candle soot reported by Mao et al.23 The cCNPs were soluble in water and formed a homogeneous, brown aqueous solution (inset, Fig. 2). An SEM study of the cCNPs showed the existence of small cCNPs with size distribution from 40 to 110 nm (Fig. 2). | ||
| Fig. 2 SEM images of cCNPs, inset: photograph of aqueous solutions of cCNPs. | ||
To characterize the morphology of cCNPs, the atomic force microscopy (AFM) and typical section analysis of cCNPs are shown in Fig. S1.† Combination of the AFM micrograph with the height profile analysis provided quantitative measurement of the dimensions of this material, indicating that cCNPs had spherical shapes with thickness distribution from 30 to 60 nm.24 About biological application of cCNPs, we estimated the size distribution of the nanoparticles in suspension from the dynamic light scattering (DLS) measurement.25 The result shows that the hydrodynamic diameters of pCNPs are broad and range from 200 to 700 nm (Fig. S2A†). For cCNPs, the diameter distribution was reduced from 40–110 nm with the majority of sizes of 70 nm (Fig. S2B†). This broad DLS sizes distribution of pCNPs and narrow size distribution of cCNPs corroborate the SEM and AFM observation. The average size of cCNPs in the DLS study is larger than those observed by SEM or AFM because DLS considers the overall hydrodynamic diameter, which includes particles as well as adsorbed molecules and ions.
We characterized the nanoparticles by Raman spectra and Fourier-transform infrared (FT-IR) spectroscopy. Fig. S3A† shows the Raman spectra of pCNPs and cCNPs, respectively using a 632.8 nm laser excitation. Two signature peaks for carbon, that is the diamondoid (D) band and graphitic (G) band,26 are clearly seen for both CNPs with the intensity ratio (ID/IG) of ∼1, indicating that the CNPs are composed mainly of the nanocrystalline graphite.27 As compared to the pCNPs, the Raman spectrum of an aqueous dispersion of the cCNPs shows an increase of the C–C tangential mode frequency at 1606 cm−1 by about 20 cm−1 with a significant intensity increase of the disorder-induced cCNPs line nears 1345 cm−1. These results reveal that the candle combustion process probably oxidized some of the chemically reactive CNPs of smaller diameter.28
The chemical groups for cCNPs were determined by analyzing the FTIR spectrum (Fig. S3B†).29 A typical FTIR spectrum of the cCNPs shows a number of infrared lines, which are assigned as follows: the line at 1620 cm−1 is assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching mode of the –COOH groups on the cCNPs, whereas the intense, broad line centered at 3405 cm−1 is assigned to the –OH stretching mode of the –COOH groups. The line at 1400 cm−1 is assigned to the CC graphitic stretching mode; these indicate existence of –COOH groups on the surface of the cCNPs.
O stretching mode of the –COOH groups on the cCNPs, whereas the intense, broad line centered at 3405 cm−1 is assigned to the –OH stretching mode of the –COOH groups. The line at 1400 cm−1 is assigned to the CC graphitic stretching mode; these indicate existence of –COOH groups on the surface of the cCNPs.
The structural compositions of the nanoparticles were obtained by estimating the atom contents from X-ray photoelectron spectroscopy (XPS) compositional analysis. For both pCNPs and cCNPs, the XPS results exhibited three main peaks of C atoms at 284.5 eV (C–C), 286.3 eV (C–O), and 287.8 eV (CO) respectively (Table S1†), suggesting that CNPs are mainly composed of graphitic carbon (sp2) and oxygen/nitrogen-bonded carbon.30 The relative amounts of various species for the pCNPs and cCNPs are summarized in Fig. S4.† It is shown that, after oxidizing the pCNPs, the relative amount of the graphitic carbon atoms on the surface of cCNPs was decreased, while the amounts of oxygen and carboxylic carbon were obviously increased. The other species of N and S showed slight increased. It could be noted that the pCNPs contained about 5.2% of carboxylic groups, which is a result of the manufacturing process by burning unscented candles. This elemental analysis is consistent with bonding structures obtained from the FT-IR spectroscopy, further demonstrating that carboxylic groups were introduced to the CNPs surface. The density of attached carboxylic groups on the cCNPs was estimated about six carbons per attached one carboxylic group. For the determination of pKa value of cCNPs, a pH titration was performed via potentiometry using commercially available glass electrodes (Fig. S5†). The measured data were processed with the program BEST,31 providing that the deprotonations of the carboxylic group of cCNPs occur at neutral or slightly alkaline condition with a pKa of 5.93 ± 0.09.
3.2. Interaction of ssDNA with cCNPs and fluorescence quenching based on electrostatic repulsion and π–π stacking
Recent work has demonstrated that ssDNA can adsorb strongly on carbon nanomaterial surfaces through π–π stacking interactions.15,16 In the case of cCNPs, however, a different behavior would be observed when the dye was linked to an oligonucleotide.21cTo examine the adsorption mechanism mentioned above, we choose P to investigate experimentally how negatively charged surface of cCNPs and ionic strength influence the interaction of the cCNPs and oligonucleotide by comparing the fluorescence quenching efficiency. Photophysical studies have found that carbon nanostructures can act collectively as quenchers for fluorophores through highly efficient long-range energy transfer from the dyes to the carbon nanostructures.32 Different from neutral organic molecules, oligonucleotides contain nitrogenous bases that are hydrophobic in nature, as well as negatively charged phosphate groups. The combination of the ionic and hydrophobic character will result in complex adsorption behavior on cCNPs surface, and the adsorption could be enhanced by screening the electrostatic repulsion with metal cations. In order to assess the effect of salts on the quenching efficiency of cCNPs for fluorescent oligonucleotides, fluorescence quenching of P by cCNPs was examined. In the presence of cCNPs, a significant decrease in the fluorescence intensity of P was observed (Fig. 3A), indicating that the ssDNA was adsorbed on the cCNPs surface. Meanwhile, with an increase in the KCl concentration, the fluorescence intensity of P was further decreased. When the concentration of KCl reached 50 mM, up to ∼97% fluorescence quenching was achieved (inset of Fig. 3A); further increase of KCl concentration, no significant changes could be observed. With a view to Mg2+ can stabilize the double helix by binding phosphate,33 we also investigated the effect of Mg2+ on the adsorption of P on cCNPs in the Tris–HCl buffer solution containing 50 mM KCl. In the presence of 10.0 mM Mg2+ ions in the buffer solution containing the same amounts of P and cCNPs, the fluorescence intensity of P was indistinguishable from the background light scattering of cCNPs with nearly 100% fluorescence quenching (Fig. 3B). The phenomena observed here support the concept that metal ions promote adsorption of ssDNA to cCNPs, and preliminarily demonstrate that binding between P and cCNPs could be improved by screening electrostatic repulsion interaction.
 | ||
| Fig. 3 (A) Fluorescence emission spectra of P (25 nM, λex = 480 nm) in 0.1 M Tris–HCl buffer solution (a), and the buffer containing 0.015 mg mL−1 cCNPs with increasing concentrations of KCl (b–g). Inset: fluorescence quenching efficiency (QE) as a function of the KCl concentration. (B) The effect of Mg2+ (100 mM) on the fluorescence quenching capability of cCNPs for P in the Tris–HCl buffer solution containing 50 mM KCl. | ||
To prove the potential application of cCNPs as a “nano-scaffold” and a “nano-quencher” for the aptamer–cCNPs assay, we investigated the fluorescence intensity change of P after adding cCNPs with different concentrations shown in Fig. 4, the fluorescence intensity decreased with the increased concentration of cCNPs, when the concentration of cCNPs solution added up to 0.1 mg mL−1, the fluorescence was quenched more than 99%, and the operation of fluorescence quenching efficiency was repeated three times. The high efficiency quenching possibly arose from strong hydrophobic adsorption of these dyes on the cCNPs surface and highly efficient long-range energy transfer from the dye to cCNPs.19 The quenching kinetics were fairly fast, with nearly 100% fluorescence quenching within only several minutes after the addition of cCNPs (Fig. S6†),which may be demonstrated to have great potential's application in ATP detection.
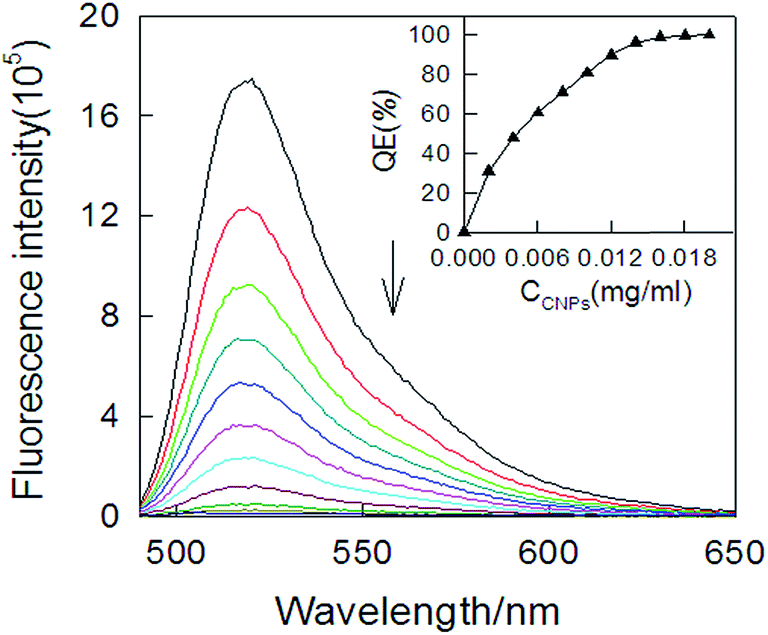 | ||
| Fig. 4 Effect of different amounts of cCNPs on the fluorescence emission spectra of P (30 nM, λex = 480 nm) in 0.1 M Tris–HCl buffer solution (pH 7.2, 100 mM KCl, 10.0 mM MgCl2). Inset: fluorescence quenching efficiency (QE%) as a function of the CNPs concentration. | ||
3.3. Fluorescence restoring of P–cCNPs complex binding to ATP
We developed a sensing platform using the noncovalent assembly of carbon nanoparticle and aptamer by fluorescence restoring of “pre-mixing” strategy for ATP detection. Fig. 5 shows the fluorescence emission spectra of P at different conditions. When adding ATP to the P–cCNPs complex solution, the fluorescence recovered rapidly. Owing to the presence of the fluorescein-based dye, the fluorescence spectrum of P (in Tris–HCl buffer) in the absence of cCNPs showed strong fluorescence emission (Fig. 5, curve a). However, in the presence of cCNPs, up to 99% quenching of the fluorescence emission were observed (Fig. 5, curve b). This observation indicated strong binding ssDNA strand to the cCNPs surface and high fluorescence quenching efficiency of cCNPs. Meanwhile, the P–cCNPs complex showed significant fluorescence enhancement by the addition of ATP (Fig. 5, curve c). The fluorescence intensity based on P–cCNPs complex changed by the addition of different concentrations of ATP. As shown in Fig. 6, the fluorescence intensity recovered gradually via concentrations of ATP from 0 μM to 1 mM, which increased approximately five-fold in the presence of 800 μM ATP, the limit of ATP detection was estimated to be about 0.12 μM, the inset of Fig. 6 illustrated the fluorescence intensity changes by the addition of different concentrations of ATP. In the presence of ATP, the phenomena of fluorescence restoring of P–cCNPs complex binding to ATP by “pre-mixing” strategy could be explained by a change of structure after single stranded aptamer binding to ATP. For the P–cCNPs complex, a dramatic increase in the fluorescence intensity was observed. Therefore, once aptamers on the cCNPs surface interacted with ATP, they would transform to the aptamer–ATP complexes. The aptamer–ATP complexes had a much lower binding ability to cCNPs and ease of disassociation from the cCNPs surface, as observed in the time-dependent experiment. As a consequence, fluorophores were far away from the cCNPs surface, and the fluorescence resonance energy transfer efficiency decreased, causing the fluorescence recovery. The “pre-mixing” strategy was proved to be sensitive to the existence of ATP and could be designed as fluorescence sensing platforms for biomolecular recognition. However, the S/B of “pre-mixing” strategy are low. Because the “pre-mixing” strategy were based on that ATP binding with aptamer had much stronger than the interaction between aptamer and cCNPs, thus made aptamer release from the surface of cCNPs, resulting in the restoration of the fluorescence. Thus, the recovery of fluorescence needed to overcome the interaction between aptamer and cCNPs. However, the “post-mixing” strategy were that dye-tagged aptamer first hybridized with ATP to form a complex, this process did not need to overcome the interaction between aptamer and cCNPs, and aptamer–ATP complexes directly formed in solution. Therefore, we favored the use of “post-mixing” strategy in the following ATP detection studies. | ||
| Fig. 5 Fluorescence emission spectra of P (30 nM) at different experiment conditions: (a) P; (b) P + cCNPs (0.015 mg mL−1); (c) P + cCNPs + 300 μM ATP; (d) P +300 μM ATP + cCNPs. | ||
 | ||
| Fig. 6 Fluorescence emission spectra of aptamer–cCNPs in the presence of different concentrations of ATP. (Inset: relative fluorescence is calculated from the fluorescence intensity ratio of S/B. The arrow indicates the signal changes as increases in the ATP concentrations. Dashed line is the fluorescence emission of P without cCNPs and ATP). | ||
3.4. Fluorescence restoring of P–ATP complex induced by cCNPs
In direct contrast to the “pre-mixing” strategy, when P was hybridized with its complementary target ATP to form a complex, the fluorescence of dye largely remained in the presence of cCNPs (Fig. 5d), suggesting the interactions between P–ATP complex and cCNPs was rather weak. These results implied that cCNPs possess significantly different adsorption affinity for P and P–ATP complex; that was P binding to cCNPs with significantly higher affinity than P–ATP. Furthermore, P bond to cCNPs in a noncovalent manner, which could be readily separated under external competition. We reasoned that this cCNPs-based fluorescence quenching might serve as a sensing platform for quantitative biomolecular analysis. Firstly, P hybridized with ATP of various concentrations for 10 min, to which an aliquot of cCNPs was added. The result demonstrated that the fluorescence of P unhybridized was efficiently quenched by cCNPs while the P–ATP complex led to observable fluorescence that formed the basis of “signal-on” aptamer sensor. As shown in Fig. 6, the fluorescence of P was intensified along with the increase of the ATP concentration.3.5. ATP detection with cCNPs aptasensor
We design a facile high-performance biosensing platforms, carbon nanoparticle aptasensor, for ATP detection using a new “post-mixing” strategy. As shown in Fig. 7, the fluorescence intensity of P and different concentrations of ATP changes made it particularly suitable for the mix-and-detection of biomolecule. At the same concentration of cCNPs, after adding cCNPs the fluorescence intensity of P largely remained. With the increasing concentration of ATP, the fluorescence intensity of P increased. Because of the extraordinarily high quenching efficiency of cCNPs, the fluorescence of the aptamer probe exhibited minimal background, while strong emission was observed when it formed aptamer–ATP complex, leading to a high signal-to-background ratio. In the presence of ATP at tenfold excess (30 nM), the S/B was increased by approximately 1.5 times as compared to that of the P (“pre-mixing” strategy). Importantly, the pre-quenched fluorescence in P–ATP complex was quickly quenched (within several minutes) in the presence of excess cCNPs and a concentration-dependent manner (Fig. 7). These results suggested that P–ATP complex had weak affinity with the cCNPs surface. This aptasensor had a good linear relationship versus ATP concentration ranging from 50 nM to 300 μM, with a detection limit of 0.1 μM ATP (Fig. S7†), which excelled “pre-mixing” strategy and ATP aptasensors (see comparison in Table S2†).34 We could obtain signals that were readily distinguishable from the background, which made it particularly suitable for mixture detection of biomolecule. | ||
| Fig. 7 The fluorescence change of the P mixing with ATP of various amounts in the presence of 0.015 mg mL−1 cCNPs. Inset: S/B of mixing ATP of various amounts. The excitation and the emission wavelengths are 490 and 520 nm, respectively. | ||
3.6. Fluorescence anisotropy
The fluorescence anisotropy of a fluorophore reflects the ability of a molecule rotate in its microenvironment.35 Anisotropy measurements are commonly used to investigate molecular interactions. As shown in Fig. S8,† based on the “pre-mixing” strategy, the fluorescence anisotropy of free P in Tris–HCl buffer was 0.064, and it increased ten-fold after addition of cCNPs, indicating that the P was adsorbed on the surface of cCNPs. However, the fluorescence anisotropy decreased by five-fold after further addition of ATP into the mixture of P and cCNPs complex, indicating the hybridization of P with ATP decreased the adsorption of P on cCNPs. Whereas basing on the “post-mixing” strategy in the absence of cCNPs, the hybridization slightly increased the fluorescence anisotropy of P, and after addition of cCNPs this value of 0.189 was lower than the value 0.341 for (4) and higher than the value 0.064 for (2). These results demonstrated the hybridization process of P–ATP complex and the adsorption of P on cCNPs for producing the FRET and fluorescence quenching, and further indicated the “post-mixing” strategy could be applied in sensitive detection of ATP.3.7. Specificity of cCNPs aptasensor
In order to evaluate the assay selectivity, a control experiment using GTP, CTP and ADP as samples were selected to study the specificity of cCNPs aptasensor. In a typical experiment, the cCNPs aptasensor was incubated with 300 μM GTP, 300 μM CTP, 300 μM ADP and 300 μM ATP in the Tris–HCl buffer, respectively. As shown in Fig. 8, no obvious signal change was observed when the cCNPs aptasensor containing GTP or CTP was incubated in the Tris–HCl buffer (data not shown), only a little increase for ADP, while a significant fluorescence increase was observed for ATP, which could be explained a lower affinity between P and GTP, CTP, ADP than that of ATP, resulting in P being adsorbed on the cCNPs surface and showing a low fluorescence. The relative fluorescence S/B of the biosensor in the presence of ATP was 6.24, which was much higher than that of 2.14 for ADP, 1.38 for GTP and 1.24 for CTP. The good selectivity of cCNPs aptasensor was attributed to the high specificity of aptamer.36 Thus, the sensor could be applied in highly sensitive detection of ATP with high specificity. The excellent biocompatibility of cCNPs also showed practical analytical application in real commercial samples.37 | ||
| Fig. 8 Relative fluorescence intensity of the cCNPs aptasensor based on the “post-mixing” strategy via a function of time incubated in blank Tris–HCl buffer; 300 μM ADP in Tris–HCl buffer; 300 μM GTP in Tris–HCl buffer; 300 μM CTP in Tris–HCl buffer; 300 μM ATP in Tris–HCl buffer, respectively. FAM–aptamer concentration: 30 nM. | ||
3.8. Application of cCNPs aptasensor
Our proposed approach for ATP assay is in a relative simple and pure buffer system. Therefore, it is necessary to investigate a further application for our assay to tolerate any interference from complex biological samples. Human urine is a very complex biological samples since consists primarily of water, with organic solutes and inorganic ions. Here, the cCNPs aptasensor is tested for quantitative detection of ATP in 20% urine solution based on the post-mixing. As shown in Fig. S9,† the fluorescence intensity of P and different concentrations of ATP changes in the presence 0.015 mg mL−1 cCNPs. The fluorescence intensity of P increases with the increasing concentration of ATP ranging from 0.25 μM to 0.8 mM, which increases approximately two-fold in the presence of 800 μM ATP. Compared to pure buffer, the approach for ATP assay has lower S/B in 20% urine solution, which indicates that our approach may suffer from some effects of urine, but because of the extraordinarily high quenching efficiency of cCNPs, as well as specific binding of ATP and aptamer, still showcase the efficiency of our proposed approach for quantification of ATP in relative complex biological fluids.4. Conclusions
Through competition of electrostatic repulsion and π–π stacking interactions cCNPs aptasensor with a “post-mixing” strategy for ATP detection, has been constructed based on the aptamer noncovalent assembly on cCNPs surfaces. The design of the present approach is simple without lengthy protocols and sophisticated probe synthesis. Meanwhile, it can be engineered in ways that offer unique advantages and capabilities that are not available from conventional molecular systems. The convenient and low-cost way for large production of cCNPs makes them ideal materials for devising biosensors. Due to the super-quenching ability of cCNPs and resulting minimization of the background fluorescence, which improve the detection sensitivity as compared to conventionally-used DNA fluorescent probes, compared to the analog designs based on “pre-mixing” strategy, the “post-mixing” strategy assay is homogeneous and shows fast hybridization kinetics, at the same time, this cCNPs aptasensor based on “post-mixing” strategy also is extraordinarily sensitive to the ATP detection with high specificity in buffer. Based on their excellent performance, the cCNPs aptasensor provides opportunities to develop simple approaches for small molecular diagnostic.Acknowledgements
The work was financially supported by the National Natural Science Foundation of China (21345006, 21275131) and the General Research Fund Scheme of University of HongKong (HKU 716809E, HKU 716310E).Notes and references
- J. R. Knowles, Annu. Rev. Biochem., 1980, 49, 877–919 CrossRef CAS PubMed.
- J. Wang, Y. X. Jiang and X. H. Fang, Anal. Chem., 2005, 77, 3542–3546 CrossRef CAS PubMed.
- K. T. Bush, S. H. Keller and S. K. Nigam, J. Clin. Invest., 2000, 106, 621–626 CrossRef CAS PubMed.
- S. Przedborski and M. Vila, Clin. Neurosci. Res., 2001, 1, 407–418 CrossRef CAS.
- R. A. Harkness and O. D. Saugstad, Scand. J. Clin. Lab. Invest., 1997, 57, 655–672 CrossRef CAS.
- Y. Eguchi, S. Shimizu and Y. Tsujimoto, Cancer Res., 1997, 57, 1835–1840 CAS.
- (a) Y. Zhang, Y. Huang and R. Yu, J. Am. Chem. Soc., 2007, 129, 15448–15449 CrossRef CAS PubMed; (b) T. Hermann and D. J. Patel, Science, 2000, 287, 820–825 CrossRef CAS; (c) M. A. Rahman, J. I. Son and Y. B. Shim, Anal. Chem., 2009, 81, 6604–6611 CrossRef CAS PubMed.
- (a) Y. F. Huang, H. T. Chang and W. Tan, Anal. Chem., 2008, 80, 567–572 CrossRef CAS PubMed; (b) I. Willner and M. Zayats, Angew. Chem., Int. Ed., 2007, 4, 6408–6418 CrossRef PubMed; (c) B. R. Baker, R. Y. Lai and K. W. Plaxco, J. Am. Chem. Soc., 2006, 128, 3138–3139 CrossRef CAS PubMed.
- (a) P. L. Sazani, R. Larralde and J. W. Szostak, J. Am. Chem. Soc., 2004, 126, 8370–8371 CrossRef CAS PubMed; (b) E. J. Merino and K. M. Weeks, J. Am. Chem. Soc., 2003, 125, 12370–12371 CrossRef CAS PubMed.
- (a) L. Cui, Y. Zou and C. J. Yang, Anal. Chem., 2012, 84, 5535–5541 CrossRef CAS PubMed; (b) J. Wang, Y. Jiang and X. Fang, Anal. Chem., 2005, 77, 3542–3546 CrossRef CAS PubMed.
- V. Bagalkot, L. Zhang and O. C. Farokhzad, Nano Lett., 2007, 7, 3065–3070 CrossRef CAS PubMed.
- B. Shlyahovsky, D. Li and I. Willner, J. Am. Chem. Soc., 2007, 129, 3814–3815 CrossRef CAS PubMed.
- L. Zhang, H. Wei and E. K. Wang, Biosens. Bioelectron., 2010, 25, 1897–1901 CrossRef CAS PubMed.
- Y. Wang, Z. H. Li and Y. H. Lin, J. Am. Chem. Soc., 2010, 132, 9274–9276 CrossRef CAS PubMed.
- R. R. Johnson, A. T. C. Johnson and M. L. Klein, Nano Lett., 2008, 8, 69–75 CrossRef CAS PubMed.
- S. W. Jung, M. Cha and J. H. Lee, J. Am. Chem. Soc., 2010, 132, 10964–10966 CrossRef CAS PubMed.
- (a) R. H. Yang, J. Y. Jin and W. H. Tan, J. Am. Chem. Soc., 2008, 130, 8351–8358 CrossRef CAS PubMed; (b) C. H. Lu, H. H. Yang and G. N. Chen, Angew. Chem., Int. Ed., 2009, 48, 4785–4787 CrossRef CAS PubMed.
- (a) B. Dubertret, M. Calme and A. J. Libchaber, Nat. Biotechnol., 2001, 19, 365–370 CrossRef CAS PubMed; (b) H. X. Li and L. J. Rothberg, Anal. Chem., 2004, 76, 5414–5417 CrossRef CAS PubMed.
- (a) R. S. Swathi and K. L. Sebastiana, J. Chem. Phys., 2008, 129, 054703 CrossRef CAS PubMed; (b) R. S. Swathi and K. L. Sebastian, J. Chem. Phys., 2009, 130, 086101 CrossRef CAS PubMed.
- (a) Q. Lu, K. O. Freedman and P. C. Ke, J. Appl. Phys., 2004, 96, 6772–6775 CrossRef CAS PubMed; (b) E. S. Jeng, A. E. Moll and M. S. Strano, Nano Lett., 2006, 6, 371–375 CrossRef CAS PubMed.
- (a) H. L. Li, J. F. Zhai and X. Sun, Biosens. Bioelectron., 2011, 26, 4656–4660 CrossRef CAS PubMed; (b) X. Ouyang, J. H. Liu and R. H. Yang, Chem. Commun., 2012, 48, 88–90 RSC; (c) J. H. Liu, J. Li and R. H. Yang, Chem. Commun., 2011, 47, 11321–11323 RSC.
- D. Eder, Chem. Rev., 2010, 110, 1348–1385 CrossRef CAS PubMed.
- H. P. Liu, T. Ye and C. D. Mao, Angew. Chem., Int. Ed., 2007, 46, 6473–6475 CrossRef CAS PubMed.
- X. R. W, S. M. Tabakman and H. J. Dai, J. Am. Chem. Soc., 2008, 130, 8152–8153 CrossRef PubMed.
- X. Liu, Q. Dai and Q. Huo, J. Am. Chem. Soc., 2008, 130, 2780–2782 CrossRef CAS PubMed.
- C. A. Furtado, U. J. Kim and E. C. Dickey, J. Am. Chem. Soc., 2004, 126, 6095–6105 CrossRef CAS PubMed.
- J. L. Dawson, M. J. Casavant and J. M. Tour, J. Am. Chem. Soc., 2004, 126, 11158–11159 CrossRef PubMed.
- A. nicaud, P. Poulin and P. Petit, J. Am. Chem. Soc., 2005, 127, 8–9 CrossRef PubMed.
- H. X. Xu and K. S. Suslick, J. Am. Chem. Soc., 2011, 133, 9148–9151 CrossRef CAS PubMed.
- Q. Shi, D. Yang and W. Yuan, J. Nanopart. Res., 2007, 9, 1205–1210 CrossRef CAS.
- A. E. Martell and R. J. Motekaitis, The determination and use of stability constants, VCH, New, York, 1988 Search PubMed.
- S. J. He, B. Song, H. P. Fang and C. H. Fan, Adv. Funct. Mater., 2010, 20, 453–459 CrossRef CAS PubMed.
- (a) Z. Zhu, Z. W. Tang and W. H. Tan, J. Am. Chem. Soc., 2008, 130, 10856–10857 CrossRef CAS PubMed; (b) H. X. Chang, L. H. Tang and J. H. Li, Anal. Chem., 2010, 82, 2341–2346 CrossRef CAS PubMed.
- (a) Y. Y. Wang and B. Liu, Analyst, 2008, 133, 1593–1598 RSC; (b) J. Huang, Z. Zhu, S. Bamrungsap, G. Z. Zhu, M. X. You, X. X. He, K. M. Wang and W. H. Tan, Anal. Chem., 2010, 82, 10158–10163 CrossRef CAS PubMed; (c) Z. A. Xu, Y. Sato, S. Nishizawa and N. Teramae, Chem.–Eur. J., 2009, 15, 10375–10378 CrossRef CAS PubMed; (d) N. Li and C. M. Ho, J. Am. Chem. Soc., 2008, 130, 2380–2381 CrossRef CAS PubMed; (e) J. Wang, L. H. Wang, X. F. Liu, Z. Q. Liang, S. P. Song, W. X. Li, G. X. Li and C. H. Fan, Adv. Mater., 2007, 19, 3943–3946 CrossRef CAS PubMed; (f) J. W. Liu, J. H. Lee and Y. Lu, Anal. Chem., 2007, 79, 4120–4125 CrossRef CAS PubMed.
- J. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 3rd edn, 2006 Search PubMed.
- A. Higuchi, Y. D. Siao and W. Y. Chen, Anal. Chem., 2008, 80, 6580–6586 CrossRef CAS PubMed.
- Y. Wang, J. Lu and J. H. Li, Anal. Chem., 2009, 81, 9710–9715 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure for the characterization of cCNPs. See DOI: 10.1039/c4ra05631k |
| This journal is © The Royal Society of Chemistry 2014 |