
Silanization of solid surfaces via mercaptopropylsilatrane: a new approach of constructing gold colloid monolayers
Wen-Hao Chena,
Yen-Ta Tsenga,
Shuchen Hsiehbc,
Wan-Chun Liua,
Chiung-Wen Hsiehb,
Chin-Wei Wua,
Chen-Han Huanga,
Hsing-Ying Lina,
Chao-Wen Chena,
Pei-Ying Linb and
Lai-Kwan Chau*a
aDepartment of Chemistry and Biochemistry and Center for Nano Bio-Detection (AIM-HI), National Chung Cheng University, Chiayi 62102, Taiwan. E-mail: chelkc@ccu.edu.tw; Fax: +886 5 2721040; Tel: +886 5 2720411 ext. 66411
bDepartment of Chemistry and Center for Biomedical Sensor Research, National Sun Yat-Sen University, Kaohsiung 804, Taiwan
cSchool of Pharmacy, College of Pharmacy, Kaohsiung Medical University, Kaohsiung 80708, Taiwan
First published on 19th September 2014
Abstract
Mercaptopropylsilatrane (MPS) was investigated as a novel self-assembled film on silica surfaces and also as a novel adhesive layer for the construction of a gold colloid monolayer on silica surfaces. We compare the preparation procedure and film quality of the MPS films to those of (3-mercaptopropyl)trimethoxysilane (MPTMS), which is commonly used for anchoring of gold nanoparticles on silica surfaces. The films were characterized by Fourier transform infrared spectroscopy, contact angle measurements, X-ray photoelectron spectroscopy, atomic force microscopy, and Ellman's reagent to determine surface mercaptan concentration. The process in preparing the MPS films involves more environmentally friendly aqueous or polar organic solvents, takes significantly shorter time (<30 min), and results in more uniform and reproducible films due to its insensitivity to moisture. The MPS films also have higher mercaptan surface density than that of the MPTMS films, resulting in higher saturation coverage of the gold colloid monolayers on the MPS-coated substrates. The higher ambient stability of the MPS films as compared to the MPTMS films is important for applications where sufficient durability of the self-assembled films under ambient conditions is required. Thus, mercaptosilatrane may have the potential to replace mercaptosilane for surface modification and as an adhesive layer for the construction of a noble metal colloid monolayer on oxide surfaces.
Introduction
Monolayers exposing the mercapto group are interesting because of their ability to coordinate to noble metals. For example, (3-mercaptopropyl)trimethoxysilane (MPTMS) is often used to construct gold colloid monolayers on silica surfaces for important applications in biosensing,1,2 surface-enhanced Raman scattering,3 catalysis,4 and so on. Unfortunately, MPTMS is very sensitive to moisture which leads to homogeneity and reproducibility problems.5–8 Although vapor deposition of MPTMS9–12 would be a good way to limit polymerization processes and prevent the effects of moisture, the reaction time is usually greater than that by liquid-phase silanization and the surface coverage is not complete.12Recently, silatranes are receiving attention from the point of view of their structural features and applications.13 Due to a strong intramolecular donor–acceptor interaction between nitrogen and silicon atoms, silatranes are chemically more stable to hydrolysis than trialkoxysilanes.14,15 As such, the low rate of hydrolysis of the silatrane molecule prevents formation of large polymer clusters on a surface.15 Hsieh and coworkers were the first to use aminopropylsilatrane as an adhesive film for anchoring gold nanoparticles (AuNPs) on silicon surfaces.16 However, the binding of amino group to gold is weaker than that of mercapto group17,18 and thus imposes limitations on the applications of fixing noble metal nanoparticles on a substrate. For example, AuNPs on amino surface can easily be displaced when the AuNP surface is further modified with a self-assembled monolayer (SAM), which is typically based the Au–S bond.
We report here a new approach to perform silanization of oxide surfaces via mercaptopropylsilatrane (MPS) to form a self-assembled film and subsequently the construction of gold colloid monolayer on the self-assembled film. To the best of our knowledge, MPS has not been used to perform silanization of oxide surfaces, nor has it been used as an adhesive film for anchoring noble metal nanoparticles on oxide surfaces. In this work, we have explored the chemistry in detail by varying solvent, concentration, and reaction time, characterized the self-assembled films using Fourier transform infrared (FTIR) spectroscopy, contact angle measurements, X-ray photoelectron spectroscopy (XPS), atomic force microscopy (AFM), and Ellman's reagent to determine surface mercaptan concentration, and examined the ambient stability of the films. Furthermore, we have explored the use of MPS as an adhesive film for anchoring gold nanoparticles on silica surfaces, and compare its performance to that of MPTMS.
Experimental
Materials
The following chemicals, (3-mercaptopropyl)trimethoxysilane (MPTMS, Sigma Aldrich), tetrachloroauric acid (Sigma Aldrich), triethanolamine (Riedel-de Haën), toluene (Macron), dichloromethane (Macron), sodium citrate (J.T. Baker), methanol (J.T. Baker), pentane (J.T. Baker), potassium carbonate (Shimakyu), sodium sulfate (Shimakyu), acetonitrile (Tedia), dimethyl sulfoxide (DMSO, Acros), and 5,5′-dithio-bis(2-nitrobenzoic acid) (DTNB, Aldrich) were used as received. All aqueous solutions were prepared with water that had been purified by using a Millipore Milli-Q water purification system (Millipore) with a specific resistance of 18.2 MΩ cm.Synthesis of MPS
MPS was prepared using a synthetic method reported earlier19 but with slight modifications. A solution of MPTMS (4.693 g, 20.3 mmol) and triethanolamine (3.2 g, 21.4 mmol) in 30 mL toluene was heated at 110 °C in a round-bottomed flask and then refluxed for about 30 h. The progress of the reaction was followed by thin layer chromatography. After cooling to room temperature, the resulting product was washed by cold n-pentane and then dried by a rotary-vacuum evaporator. The dried powder was re-dissolved in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dichloromethane/n-pentane. After re-crystallization twice, a white solid 2.30 g MPS (9.2 mmol) with a yield of 45.5% was obtained. The deposit was re-dissolved in DMSO (20 mL) as a stock solution for storage.
:1 dichloromethane/n-pentane. After re-crystallization twice, a white solid 2.30 g MPS (9.2 mmol) with a yield of 45.5% was obtained. The deposit was re-dissolved in DMSO (20 mL) as a stock solution for storage.
Fourier transform infrared spectroscopy (FTIR) and hydrogen nuclear magnetic resonance (1H NMR) were used to examine the purity and structure. 1H 200 MHz NMR (CDCl3): 0.285 (SiCH2, 2H, t, J = 16 Hz), 1.22 (HS, 1H, t, J = 12 Hz), 1.55 (CH2, 2H, m, J = 42.6 Hz), 2.64 (SCH2, 2H, t, J = 16 Hz), 2.788 (OCH2, 6H, t, J = 11.2 Hz), 3.647 (NCH2, 6H, t, J = 11.2 Hz). FTIR (KBr, cm−1): 2928 and 2876 (vs., νCH2), 2545 (w, νSH), 1454 and 1277 (m, δCH2), 1124 and 1100 (vs., νCO), 939 (s, νCC), 909 (s, νCN), 776 (s, ν(as)SiO), 616 (m, ν(s)SiC), and 582 (m, νN→Si).14,19
Preparation of AuNPs
AuNPs were synthesized according to the following procedure. HAuCl4·3H2O (0.88 mM, 20 mL) was heated to boiling with vigorous stirring. A fresh 1% sodium citrate solution (2.4 mL) was added in the boiling solution rapidly. The color of the solution turned from yellow to colorless, then very dark purple, then ruby-red. The resulting solution was kept boiling for 20 min, then cooled down to room temperature and stored at 4 °C as a stock solution. The stock AuNP solution was characterized by a UV-visible spectrometer. Typically, the peak wavelength was at about 520 nm and the peak absorbance was about 2.6. For preparation of gold colloid monolayers, the stock AuNP solution was diluted by 1% sodium citrate solution until the absorbance was 2.0. As such, the concentration of the diluted AuNP solution is about 10 nM.20 Histograms of the images derived from transmission electron microscopy (TEM) revealed that the mean diameter of the AuNPs was 13 ± 1 nm.Preparation of mercaptan-functionalized substrates
Glass slides with dimensions 5.5 cm × 0.8 cm were cleaned by soap and then sonicated sequentially in methanol, acetone, purified water, methanol, acetone, and purified water, with sonication time of 10 min in each step. The glass slides were dried in an oven at 70 °C and then stored at room temperature. Before use, the glass slides were treated by oxygen plasma (0.6 torr, Harrick Scientific Products, Inc.) for 20 min.The MPTMS-modified substrates were prepared as follows. A solution of 5% MPTMS in toluene (v/v, about 0.25 M) was allowed to prehydrolysis for 12 h. Then the glass slides were immersed into a 5% MPTMS solution for 12 h. Subsequently, the modified glass slides were sonicated sequentially in methanol–toluene (1:1, v/v), methanol, and purified water for 15 min in each step, and then dried under a nitrogen stream.
The MPS-modified substrates were prepared as follows. The stock MPS solution was diluted to 1:1000 (v/v, about 0.46 mM) in methanol and the glass slides were immersed into the solution for 30 min. Subsequently, the modified glass slides were rinsed by methanol and purified water and then dried by nitrogen stream.
Quantification of mercaptan surface density
Samples of MPS and MPTMS films on glass slides were immersed in a cuvette containing 1 mL of Ellman's reagent, (4 μM DTNB in 0.1 M pH 8.0 phosphate buffer). The reaction was allowed to proceed for 12 h at room temperature and the absorbance was measured at the wavelength of 412 nm. Controlled samples were elaborated with non-modified glass slides. The amount of mercaptan moieties was calculated from the corresponding standard curve elaborated between 2 × 10−8 M and 5 × 10−6 M of mercaptoethanol.Ambient stability test
The modification of the substrates by MPTMS (toluene 16 h) or MPS (MeOH 30 min) was under dark and room temperature conditions according to the procedures as described above. Once a substrate had finished the modification procedures, the stability of the modified substrate is tested immediately under room light and room temperature conditions for standing time periods of 0, 1, 3, 24, and 48 h. Then the modified substrate was immersed in a diluted AuNP solution to anchor AnNPs on the modified surface. The result of the stability test was observed by measuring the UV-visible spectra of the AuNPs anchored on the modified substrate.Instrumentation
UV-visible absorption spectra were obtained with full spectrum from 400 to 800 nm by a Jasco V-570 spectrometer. TEM images were recorded using a JEOL 2010 HRTEM operating at an accelerating voltage of 200 kV. Scanning electron microscopy (SEM) images for determination of the coverage of the gold colloid monolayers were obtained by a Hitachi 4800I field-emission scanning electron microscope (FE-SEM) operated at 15 kV. The surface morphology of the films was characterized using an atomic force microscope (MFP-3D™, Asylum Research, Santa Barbara, CA) under ambient conditions. For AFM experiments, a silicon cantilever (Olympus AC240TS) with a normal spring constant of 2 N m−1 (thermal method) (Proksch, Schaffer, Cleveland, Callahan, & Viani, 2004) was used. All AFM images were acquired in AC (tapping) mode with an image resolution of 512 × 512 pixels.Fourier transform infrared (FTIR) spectroscopy in the transmission mode was used to characterize the bulk MPTMS and MPS samples and the molecular packing of the MPTMS and MPS films. To collect IR spectra of bulk samples, MPTMS or MPS was casted onto a KBr pellet and FTIR spectra was recorded on a Bruker Alpha FTIR spectrometer (4 cm−1 resolution, 2048 scans) equipped with a deuterated triglycine sulfate (DTGS) detector. To collect IR spectra of thin films, a MPTMS film or a MPS film was prepared on a silicon wafer and FTIR spectra was recorded on a Bruker 66 v/s FTIR spectrometer equipped with a liquid nitrogen-cooled mercury–cadmium–telluride (MCT) detector (8 cm−1 resolution, 256 scans, sample compartment vacuum pressure was 0.12 hPa).
Equilibrium water contact angles were measured on modified silicon wafer surfaces and on a bare silicon wafer substrate using a CAM-M200 contact angle meter (Creating Nano Technologies, Inc., Taiwan) at room temperature. All the contact angles measured in this study were undertaken with 10 μL volume at different locations on each sample surface.
XPS was measured with a Kratos Axis Ultra DLD spectrometer using a Mg/Al achromatic source 450 W. Each sample was analyzed at photoelectron take-off angle of 45°. The take-off angle is defined as the angle between the analyzer and the substrate surface. The silica substrate was used an internal standard for XPS positions and a binding energy of 99.15 eV was used for elemental silicon.21 The peak deconvolution was performed after baseline subtraction by assuming that the constituent peaks have Gaussian shape.
Results and discussion
The mercapto group is known to have specific interaction with many metals, including gold, silver, and copper. For this purpose, MPTMS is one of the popular precursors to prepare a mercaptan-functionalized surface for anchoring of noble metal nanoparticles. Here, we explore the use of MPS to prepare mercaptan-functionalized surfaces and compare its performance to that of MPTMS. The procedures used to prepare MPS- or MPTMS-modified surfaces are illustrated schematically in the first step shown in Fig. 1. | ||
| Fig. 1 Schematic illustration showing the procedures for surface functionalization by MPTMS or MPS and anchoring of AuNPs on those functionalized substrates. | ||
Literature review shows that the MPTMS system usually uses a concentration of 0.1% v/v–5% v/v and takes 0.5 h–24 h in anhydrous organic solvent, such as toluene and benzene, to form a well-ordered film.5,6,21 However, MPTMS is very sensitive to moisture which leads to agglomeration and polymerization on a substrate during deposition, generally resulting in multilayer deposition and irregular surface morphology.5–8 On the other hand, silatranes are relatively stable to moisture13,16 and thus may be applicable in more polar solvents. In this aspect, using the capability of the films to anchor AuNPs as an indicator to monitor the film formation process, we found that films formed from MPS dissolved in nonpolar solvents (e.g., toluene) or polar solvent (e.g., methanol) have similar activity and kinetics (data not shown). Surprisingly, an aliquot of stock MPS solution diluted by water also show good activity towards anchoring of AuNPs. The films were then characterized using FTIR spectroscopy, contact angle measurements, XPS, AFM, and Ellman's reagent.
Infrared spectroscopy
FTIR spectroscopy was performed to confirm the presence of the MPS and MPTMS films on silicon wafer samples and evaluate their structures. Fig. 2 shows the spectra in the region between 700 and 3300 cm−1 that contain the vibrational modes of CH2 stretching, CH3 stretching, SH stretching, and Si–O–Si stretching. Fig. 2a illustrates the IR spectrum of bulk MPS with peaks of 2928, 2876 cm−1 for asymmetric and symmetric CH2 stretching, and 2545 cm−1 for SH stretching, respectively. A characteristic peak is also found at 582 cm−1 (data not shown), which has been assigned to the vibration of the coordination bond (N → Si) in silatranes.14,19 For the IR spectrum of the MPS film shown in Fig. 2a, we have observed CH2 stretching modes at 2927 and 2860 cm−1, and SH stretching mode at 2545 cm−1. These peak positions and assignments are in agreement with literature.19,22 Comparatively, the IR spectrum of bulk MPTMS shown in Fig. 2b indicates peaks at 2942 cm−1 for asymmetric CH3 stretching and 2841 cm−1 for symmetric CH3 stretching of the methoxyl groups, and 2565 cm−1 for SH stretching.23–25 The IR spectrum of the MPTMS film shown in Fig. 2b indicates CH2 stretching modes at 2927 and 2870 cm−1, and SH stretching mode at 2545 cm−1. The disappearance of the CH3 stretching modes suggests that hydrolysis and condensation occurred within the MPTMS film. | ||
| Fig. 2 FTIR spectra in the 3300−2200 cm−1 range for (a) the MPS film and bulk and (b) the MPTMS film and bulk; (c) FTIR spectra in the 1400−700 cm−1 range for the MPS and MPTMS films. | ||
The positions of the peak frequencies for CH2 stretching, SH stretching, and Si–O–Si stretching modes provide insight into the intermolecular environment in these films.26,27 First, the relatively high CH2 vibrational frequencies suggest that the propyl chains in both the MPS and MPTMS films are more liquid-like. On the other hand, the lower CH2 symmetric stretching frequency for the MPS film at 2860 cm−1 as compared to that of MPTMS film at 2870 cm−1 suggests that the self-assembled MPS molecules have a better packing in the film. Second, it has been reported that the SH stretching mode free from hydrogen bonding is at 2565 cm−1 while SH involved in hydrogen bonding has the SH stretching mode at lower frequency.25,28,29 As such, it suggests that the SH group in bulk MPTMS is free from hydrogen bonding while the SH group in the MPTMS film, bulk MPS, and MPS film probably has significant SH⋯S interaction.28,29 Third, peak positions and widths of the Si–O–Si vibrational modes in the 1400−400 cm−1 range reflect the structural orderedness of the silica skeleton.30,31 By considering long-range interaction provided by the Coulomb fields in the silica skeleton, Galeener and Lucovsky have accounted for three pairs of longitudinal optical (LO) and transverse optical (TO) vibrational modes in the vibrational spectra of vitreous silica.30 As shown in Fig. 2c, the IR spectra in the 1400−700 cm−1 range show that the MPS film has two peaks at 1136 and 1073 cm−1, which are respectively assigned to the LO and TO modes of the Si–O–Si asymmetric stretching vibration, while the MPTMS film has only one peak at 1037 cm−1. The LO–TO splitting exhibited in the IR spectrum of the MPS film suggests that the silica skeleton in the MPS film is sufficiently in order.
Contact angle measurements
To further characterize the films, water contact angle measurements were performed on the surfaces of the MPS- and MPTMS-modified silicon wafer samples. The contact angle for ethanol cleaned silicon wafers was 24.6° ± 2.3°, which is indicative of a hydrophilic surface. The contact angle for the MPS and MPTMS films was much higher, at 57.3° ± 1.1° and, 57.3° ± 4.2°, respectively. Such values are consistent with values reported in literature21,24,32,33 and further confirm the presence of the respective films on the silicon surfaces. The better reproducibility of the contact angle for the MPS film also suggests that the MPS film is more uniform than the MPTMS film.X-ray photoelectron spectroscopy
Fig. 3a shows representative XPS spectra of a MPS film (line I) and a MPTMS film (line II) on glass substrate. Both spectra are very similar and confirm the presence of four elements (Si, C, O and S) in the films. Such a spectral similarity suggests that the atomic composition of the end product of both films is similar. The most significant feature in the XPS spectrum of the MPS film is the absence of N signal (∼400 eV), which agrees with a proposed mechanism of partial hydrolysis of the silatrane followed by the reaction with a silica surface15 and our proposed mechanism as shown in Fig. 4. The absence of the N peak also confirms that, after the hydrolysis, no triethanolamine was left as a byproduct on the substrate. | ||
| Fig. 3 (a) The full XPS spectra, (b) the C(1s) XPS spectra, and (c) the S(2p) XPS spectra of a MPS film (line I) and a MPTMS film (line II). Curve-fitting of the C(1s) XPS spectra and the S(2p) XPS spectra are also shown. | ||
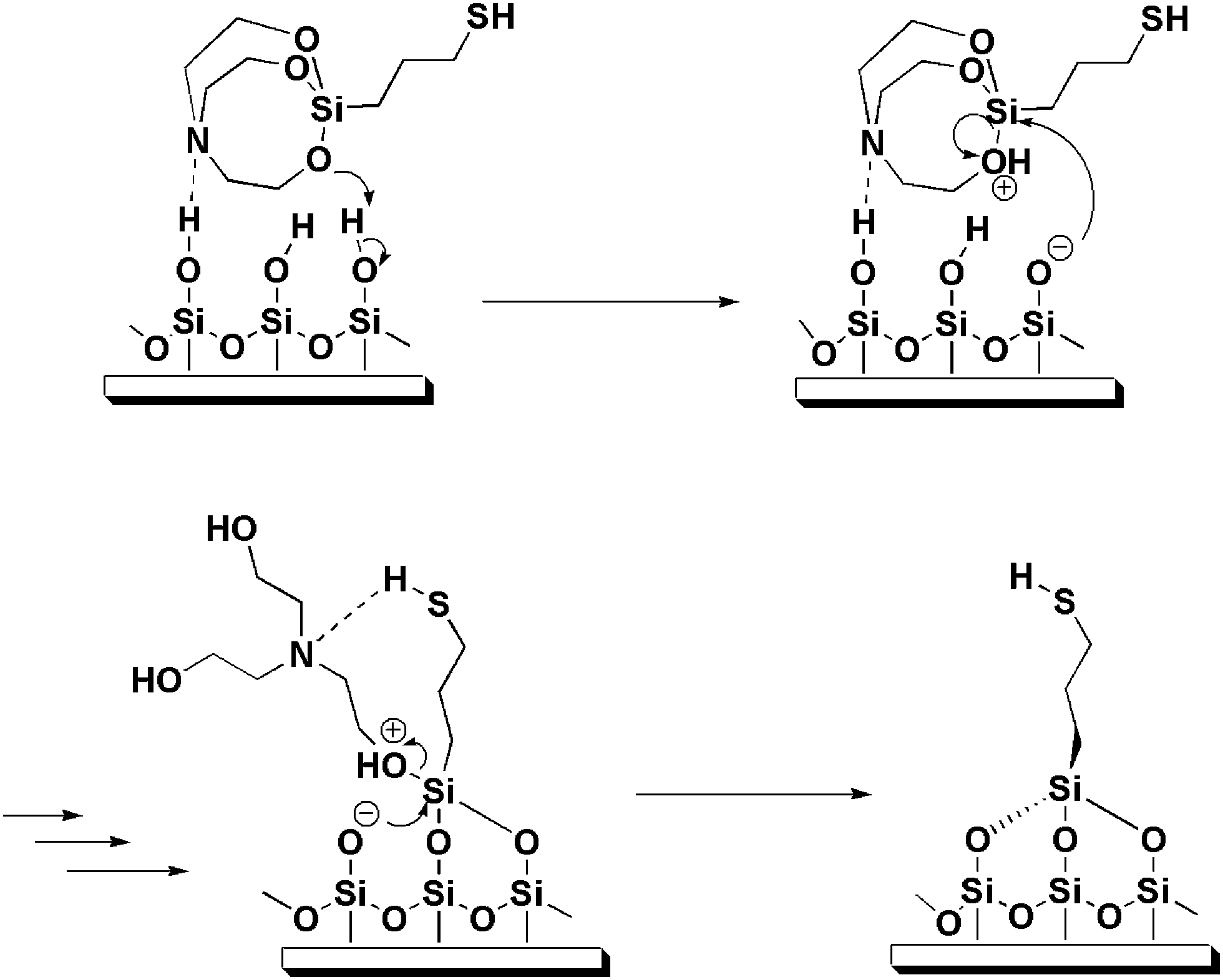 | ||
| Fig. 4 Schematic representation of the mechanism proposed for the reaction between MPS and surface silanol. | ||
Fig. 3b shows XPS C(1s) spectra of the MPS film (line I) and the MPTMS film (line II) on glass substrate. The C(1s) XPS spectrum of bare silica substrate shows only one band at 284.7 eV, whereas those of the MPS and MPTMS films on silica substrate show two contribution bands at 284.7 eV and 286.3 eV. The band at 284.7 eV corresponds to hydrocarbon and carbon bonded to silicon and the band at 286.3 eV corresponds to carbon bonded to oxygen and sulfur.5 The appearance of a new band at 286.3 eV demonstrates indeed both MPS and MPTMS react with the SiO2 surface.
Fig. 3c shows XPS S(2p) spectra of the MPS film (line I) and the MPTMS film (line II) on glass substrate. The S(2p) core-level band of both MPTMS and MPS films can be decomposed into three peaks at binding energies of 162.1, 165.4, 169.5 eV and 162.1, 165.5, 169.6 eV, respectively. Comparison of the area integration of the sulfur peaks for MPS:MPTMS is 1.42:1, suggesting that the MPS film has a higher surface sulfur density than that of the MPTMS film. This is consistent with the FTIR results where the MPS modified substrate shows a larger SH stretching peak at 2545 cm−1. The S(2p3/2,1/2) doublet feature of both films at 162.1/165.4 eV and 162.1/165.5 eV are attributed to either reduced sulfur (–SH) or to disulfide (–SS–) species.32,34 XPS alone cannot distinguish between the different kinds of reduced sulfur, mercaptan, or disulfide.34 The other peak at 169.5 eV or 169.6 eV is consistent with oxidized sulfur (–SOxH).32,35,36 The area ratios of the oxidized sulfur band with respect to the total sulfur curve for the MPS film and the MPTMS film are 56.1% and 74.2%, respectively, suggesting that the mercapto group in the MPS film is less susceptible to oxidation. It should be noted that oxidation may take place during storage of the sample until XPS measurement37 and X-rays can have a damaging effect on the surface structures.38 Nevertheless, considering that both films were prepared and analyzed at the same time, these two factors should affect both films to the same extent.
Quantification of mercaptan surface density
Surface density of the mercapto group is of great concern, because it determines the density of self-assembled molecules on top of the films and hence the reactivity of the films toward anchoring of AuNPs. The Ellman's reaction was first developed for the determination of mercapto group in biological materials39 and later on also applied to the determination of surface mercapto group.40 In this study, quantification of the number of accessible mercapto group of the MPS and MPTMS films was performed by using the Ellman's reagent, 5,5′-dithio-bis(2-nitrobenzoic acid) (DTNB). The optimum reaction time between the films and DTNB was investigated by monitoring the absorbance at 412 nm over a time period of 2 to 12 h. Beyond 10 h of reaction time, the absorbance value level off and hence a reaction time of 12 h was used for subsequent analyses. The mean mercaptan surface density of the MPS and MPTMS films was 5.4 ± 0.4 nm2 and 4.1 ± 0.4 nm2, respectively. The mercaptan surface density of the MPS film is consistent with the average number of hydroxyl group of ∼5 nm−2 typically found on silica surfaces41,42 and is also reasonable as compared to a total of about 8 surface Si atom per nm2.43 Furthermore, the estimated mercaptan surface density agrees with the approximate area (0.20–0.21 nm2) projected by each alkyl chain in the plane of close-packed alkylsiloxane monolayers.44,45 In addition, the higher mercaptan surface density of the MPS film as compared to that of the MPTMS film is consistent with the XPS and FTIR results, suggesting that the self-assembled MPS molecules have a better packing density in the film and/or the mercapto group in the MPS film is less susceptible to oxidation.Atomic force microscopy
Fig. 5a and b shows a typical series of AFM 2D and 3D images displaying the surface of a MPTMS-modified silicon substrate. The size of the AFM scans is 1 × 1 μm2. The images show the surface morphology of the MPTMS film, a few of small protrusions appear on the surface. The bright spots in the image indicate where aggregates have formed. Such an observation is consistent with previous reports and is attributed to the competition between self-polymerization and condensation reaction with surface silanol.5,8 The surface roughness is 137 pm as calculated from Fig. 5a. Fig. 5c and d show a typical series of AFM 2D and 3D images displaying the surface of a MPS-modified silicon substrate. The surface morphology of the MPS-modified silicon substrate shows that the film is homogeneous and smooth. The calculated surface roughness is 110 pm. The AFM results show that the MPS film is smoother than the MPTMS film. This is consistent with the better reproducibility of contact angle measurement for the MPS modified surface. The higher roughness of the MPTMS film has been attributed to unhydrolyzed Si–OCH3 bonds which make lateral cross-linking between neighbouring adsorbed MPTMS molecules more difficult.8 Incomplete lateral cross-linking may result in a lower surface coverage and such a proposition is consistent with the results of surface mercaptan concentration determination. | ||
| Fig. 5 AFM 2D and 3D images. (a) 3D image and (b) 2D image of MPTMS modified silicon surface (0.25 M in toluene, 12 h). (c) 3D image and (d) 2D image of MPS modified silicon surface (0.46 mM in methanol, 30 min). | ||
Kinetics of film formation
To investigate the formation kinetics of both the MPS and MPTMS films, the high-extinction surface plasmon resonance (SPR) of gold colloid allows us to monitor the film formation process by following the UV-vis spectra of the anchored particles with increasing film formation time. In this study, the absorbance of two sets of glass slides was measured after the glass slides in each set were immersed separately into a MPS solution or a MPTMS solution for various time periods. The MPTMS- and MPS-coated glass slides were then immersed in a 10 nM AuNP solution for 30 min (see below). Fig. 6 shows the trends of absorbance at 530 nm versus immersion time for the two set of glass slides. As shown in previous studies46,47 and in this study (curve a of Fig. 6), MPTMS needs a long time (>8 h) to form a complete layer with optimum mercaptan surface density. In contrast, as shown in curve b of Fig. 6, MPS requires just 25 min to form a complete layer with optimum mercaptan surface density. The faster formation rate of the MPS film is probably due to our proposed mechanism as shown in Fig. 4. First, there is likely an attractive interaction between the electron lone pair on the N atom of the silatrane and the silanol group on the silica surface so that silatrane molecules reach the silica surface much faster. Second, ab initio calculations show that the transannular Si–N interaction leads to a larger proton affinity of the oxygen atoms in silatranes than that in trimethoxysilanes and thus facilitates the condensation reaction at the silica surface.48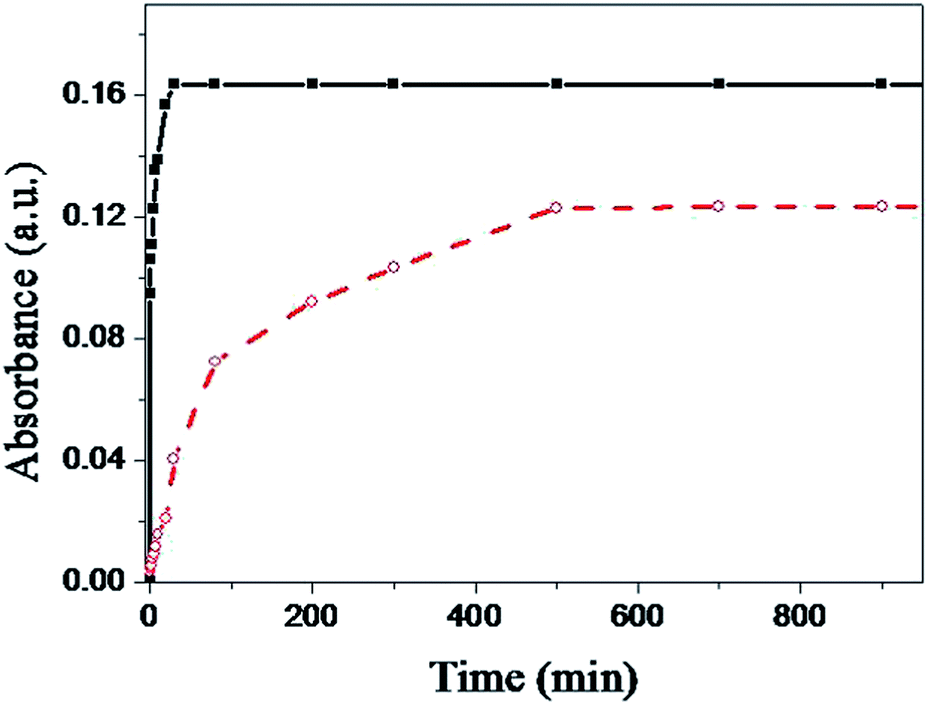 | ||
| Fig. 6 Absorbance at 530 nm versus immersion time for two sets of glass slides with each set of glass slides immersed separately into a solution of (■) MPS (0.46 mM in MeOH) or (○) MPTMS (0.25 M in toluene) and then immersed in a 10 nM AuNP solution for 30 min. | ||
Anchoring gold nanoparticles
Many processes are involved in the formation of a gold colloid film on mercaptan-functionalized surfaces. Natan and coworkers found that gold colloid monolayer coverage is limited at early times by diffusion and at later times by interparticle repulsion.46 When the MPTMS- and MPS-coated glass slides were immersed in a solution of 13 nm diameter AuNPs (AuNP concentration = 10 nM, absorbance = ∼2.0 A.U.) with various immersion times, we found that both the MPTMS- and MPS-modified substrates began to reach the saturation coverage at about 30 min (data not shown). Hence, we used a reaction time of 30 min for preparation of gold colloid films.Fig. 7 shows the typical UV-vis spectra and FE-SEM images of the gold colloid monolayer on a MPTMS-coated substrate and a MPS-coated substrate prepared at optimized conditions. As can be seen in the FE-SEM images, the mean diameter of the AuNPs is still about 13 nm and particle aggregation is almost not present in carefully prepared surfaces. Similar conclusions are also obtained by AFM (data not shown). It should be noted that the saturation coverage of the gold colloid monolayer on the MPS-coated substrates (26.5%) is higher that on the MPTMS-coated substrates (22.6%), while the full-width half-maximum of the SPR band is similar (82.1 nm versus 93.2 nm). Since gold colloids possess mobility and might aggregate to larger colloids, UV-vis spectra and FE-SEM images of the gold colloid monolayer after longer period of storage time were traced. In a period of about 2 months, no significant difference in UV-vis spectra and FE-SEM images were observed. As AuNPs are important sensor materials today, reproducibility of the sensor fabrication process is important in order to meet analytical performance standards. In this regard, we explore the reproducibility of using both MPTMS and MPS as linkers to prepare gold colloid monolayers on glass slides. Results show that the average coefficients of variation (CV) of the peak absorbance for batch-to-batch runs (n = 7) with MPTMS and MPS are 2.3% and 1.6%, respectively, while the coefficients of variation (CV) of the peak absorbance for day-to-day runs (n = 7) with MPTMS and MPS are 5.7% and 3.4%, respectively. Although the reproducibility of using MPTMS as a linker is acceptable, the reproducibility of using MPS as a linker is even better. Together with the shorter preparation time required, we believe MPS will benefit the development of sensors based on gold colloid technology.
 | ||
| Fig. 7 Absorbance spectra and FE-SEM images of AuNPs on a MPS-coated glass slide (line I) and a MPTMS-coated glass slide (line II). | ||
Ambient stability
The ultimate utility of mercaptan-functionalized films will be critically dependent on their stability under ambient conditions. Organothiol monolayers on Au exposed to air for prolonged periods has been shown to be oxidized to sulfinates and sulfonates, indicating their finite stability.37,49–51 To explore the stability of MPTMS and MPS films under ambient laboratory conditions, the MPTMS- and MPS-coated glass slides were exposed in ambient air under room light at various standing times and then immersed in a solution of AuNPs for 30 min. Then we define the films with negligible exposure to ambient air under room light (i.e., zero standing time) have 100% activity. In other words, activity is defined as Ai/A0 where A0 is the absorbance at zero standing time, Ai is the absorbance at a finite standing time, and the absorbance is measured at 530 nm.As shown in Fig. 8, the activity of the MPTMS films at a standing time of 1 h remains 20%, while the activity at a standing time of 25 h remains just 10%. On the contrary, there is not observable loss in activity for the MPS films at a standing time of 1 h. At a standing time of 25 h, the activity of the MPS films remains 82%, while the activity at a standing time of 48 h still remains more than 75%. This is not a trivial result as the end products of both films are expected to be similar. Senkevich and coworkers have demonstrated that the mercapto group in the starting MPTMS molecule is stable in liquid form over a period of 8 months, suggesting that the oxidized mercaptan molecular layer is not due to the oxidation of the starting molecule but rather due to the chemistry residing on the surface.34 It is well-known that the hydrolysis of the MPTMS molecule in the film form is often incomplete and thus a MPTMS film often retains its methoxy groups.21 As the pKa of propyl mercaptan (pKa = 10.6)52 is lower than that of water (pKa = 15.7), we propose a reaction mechanism as shown in Fig. 9 to account for the lower ambient stability of the MPTMS film. Since the mercapto group is more acidic than water molecules from moisture, it will accelerate the hydrolysis of a neighbouring methoxy group in a MPTMS film and form a thiolate itself. The thiolate thus formed is susceptible to oxidation,53 for example, reacts with oxygen to form sulfenic acid. Sulfenic acid may then be further oxidized to sulfinic acid and sulfonic acid. On the contrary, the MPS film is relatively stable because of the complete hydrolysis of the Si–O–C bonds in the silatrane, as suggested by the XPS results.
 | ||
| Fig. 8 Activity of the (■) MPS and (○) MPTMS films after exposure in ambient air under room light at various standing times (n = 3). | ||
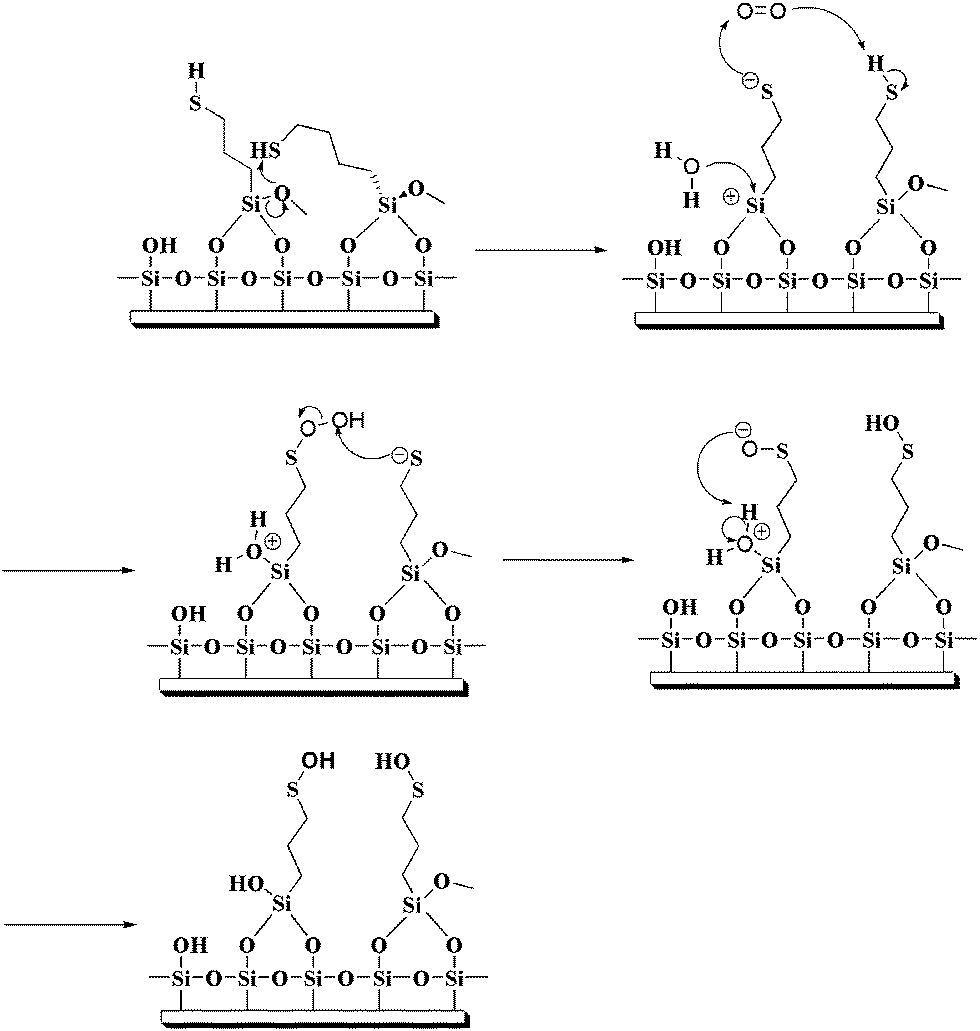 | ||
| Fig. 9 Schematic representation of the mechanism proposed for the oxidation of the mercapto group in MPTMS films. | ||
Conclusions
In this study, we have shown that MPS can be used to form a self-assembled film on silica surfaces and also as an effective adhesive layer for the construction of a gold colloid monolayer on silica surfaces. As silatranes are relatively stable to moisture, MPS is applicable in more environmentally friendly aqueous and polar organic solvents while MPTMS usually works better in anhydrous organic solvents. In comparison with MPTMS, the MPS films are smoother and more uniform, and are completely free of molecular aggregates. MPS also requires a significantly shorter time than MPTMS to form a complete layer and exhibits a higher mercaptan surface density in the film. Such a higher mercaptan surface density in the MPS film facilitates the anchoring of gold nanoparticles on the film. As a result, the saturation coverage of the gold colloid monolayer on the MPS-coated substrates is higher that on the MPTMS-coated substrates. Surprisingly, the MPS films also exhibit a higher ambient stability as compared to the MPTMS films. This characteristic is beneficial to many applications where sufficient durability of the self-assembled films under ambient conditions is important. Thus, mercaptosilatrane as an efficient and environmental-amiable molecular linker may have a potential to replace mercaptosilane for surface modification.Acknowledgements
This work was supported by Ministry of Science and Technology (Taiwan) through grant number NSC 102-2113-M-194-003-MY3.References
- From Bioimaging to Biosensors - Noble Metal Nanoparticles in Biodetection, ed. L.-K. Chau and H.-T. Chang, Pan Stanford Publishing, Singapore, 2013 Search PubMed
.
- B. Sepulveda, P. C. Angelome, L. M. Lechuga and L. M. Liz-Marzan, Nano Today, 2009, 4, 244–251 CrossRef CAS
- R. G. Freeman, K. C. Grabar, K. J. Allison, R. M. Bright, J. A. Davis, A. P. Guthrie, M. B. Hommer, M. A. Jackson, P. C. Smith, D. G. Walter and M. J. Natan, Science, 1995, 267, 1629–1632 CrossRef CAS PubMed
- D. T. Thompson, Nano Today, 2007, 2, 40–43 CrossRef
- M. Hu, S. Noda, T. Okubo, Y. Yamaguchi and H. Komiyama, Appl. Surf. Sci., 2001, 181, 307–316 CrossRef CAS
- C. M. Halliwell and A. E. G. Cass, Anal. Chem., 2001, 73, 2476–2483 CrossRef CAS PubMed
- A. V. Krasnoslobodtsev and S. N. Smirnov, Langmuir, 2002, 18, 3181–3184 CrossRef CAS
- S.-R. Yang and B. O. Kolbesen, Langmuir, 2008, 255, 1726–1735 CAS
- E. Pavlovic, A. Quist, U. Gelius and S. Oscarsson, J. Colloid Interface Sci., 2002, 254, 200–203 CrossRef CAS PubMed
- P. Doppelt, N. Semaltianos, C. D. Cavellin, J. L. Pastol and D. Ballutaud, Microelectron. Eng., 2004, 76, 113–118 CrossRef CAS
- E. Finocchio, E. Macis, R. Raiteri and G. Busca, Langmuir, 2007, 23, 2505–2509 CrossRef CAS PubMed
- B. Pattier, J.-F. Bardeau, M. Edely, A. Gibaud and N. Delorme, Langmuir, 2008, 24, 821–825 CrossRef CAS PubMed
- J. K. Puri, R. Singh and V. K. Chahal, Chem. Soc. Rev., 2011, 40, 1791–1840 RSC
- M. G. Voronkov, Pure Appl. Chem., 1966, 13, 35–60 CrossRef CAS
- L. S. Shlyakhtenko, A. A. Gall, A. Filonov, Z. Cerovac, A. Lushnikov and Y. L. Lyubchenko, Ultramicroscopy, 2003, 97, 279–287 CrossRef CAS PubMed
- S. Hsieh, W.-J. Chao and C.-W. Hsieh, J. Nanosci. Nanotechnol., 2009, 9, 2894–2901 CrossRef CAS PubMed
- C. A. Martin, D. Ding, H. S. van der Zant and J. M. van Ruitenbeek, New J. Phys., 2008, 10, 065008 CrossRef
- D. V. Leff, L. Brandt and J. R. Heath, Langmuir, 1996, 12, 4723–4730 CrossRef CAS
- R. Spennato, M.-J. Menu, M. Dartiguenave and Y. Dartiguenave, Transition Met. Chem., 2004, 29, 830–839 CrossRef CAS
- X. Liu, M. Atwater, J. Wang and Q. Huo, Colloids Surf., B, 2007, 58, 3–7 CrossRef CAS PubMed
- J. J. Senkevich, G.-R. Yang and T.-M. Lu, Colloids Surf., A, 2002, 207, 139–145 CrossRef CAS
- Y. Yu, J. Addai-Mensah and D. Losic, J. Nanosci. Nanotechnol., 2011, 11, 1–8 CrossRef CAS
- Y.-S. Li, Y. Wang, T. Tran and A. Perkins, Spectrochim. Acta, Part A, 2005, 61, 3032–3037 CrossRef PubMed
- D. G. Kurth and T. Bein, Langmuir, 1993, 9, 2965–2973 CrossRef CAS
- M. Hayashi, Y. Shiro and H. Murata, Bull. Chem. Soc. Jpn., 1966, 39, 112–117 CrossRef CAS
- R. G. Snyder, H. L. Strauss and C. Elliger, J. Phys. Chem., 1982, 86, 5145–5150 CrossRef CAS
- M. D. Porter, T. B. Bright, D. L. Allara and C. E. D. Chidsey, J. Am. Chem. Soc., 1987, 109, 3559–3568 CrossRef CAS
- B. A. Kolesov, V. S. Minkov, E. V. Boldyreva and T. N. Drebushchak, J. Phys. Chem. B, 2008, 112, 12827–12839 CrossRef CAS PubMed
- V. S. Minkov, S. V. Goryainov, E. V. Boldyreva and C. H. Gorbitz, J. Raman Spectrosc., 2010, 41, 1748–1758 CrossRef
- F. L. Galeener and G. Lucovsky, Phys. Rev. Lett., 1976, 37, 1474–1478 CrossRef CAS
- R. B. Laughlin and J. D. Joannopoulos, Phys. Rev. B: Solid State, 1977, 16, 2942–2952 CrossRef CAS
- J. Liu and V. Hlady, Colloids Surf., B, 1996, 8, 25–37 CrossRef CAS
- J. J. Cras, C. A. Rowe-Taitt, D. A. Nivens and F. S. Ligler, Biosens. Bioelectron., 1999, 14, 683–688 CrossRef CAS
- J. J. Senkevich, C. J. Mitchell, G.-R. Yang and T.-M. Lu, Langmuir, 2002, 18, 1587–1594 CrossRef CAS
- N. Balachander and C. N. Sukenik, Langmuir, 1990, 6, 1621–1627 CrossRef CAS
- N. G. Semaltianos, Surf. Coat. Technol., 2007, 201, 7327–7338 CrossRef CAS
- T. M. Willey, A. L. Vance, T. van Buuren, C. Bostedt, L. J. Terminello and C. S. Fadley, Surf. Sci., 2005, 576, 188–196 CrossRef CAS
- K. Heister, M. Zharnikov, M. Grunze, L. S. O. Johansson and A. Ulman, Langmuir, 2001, 17, 8–11 CrossRef CAS
- G. L. Ellman, Arch. Biochem. Biophys., 1959, 82, 70–77 CrossRef CAS PubMed
- J. D. Bass and A. Katz, Chem. Mater., 2006, 18, 1611–1620 CrossRef CAS
- L. T. Zhuravlev, Langmuir, 1987, 3, 316–318 CrossRef CAS
- A. Tuel, H. Hommel and A. P. Legrand, Langmuir, 1990, 6, 770–775 CrossRef CAS
- M. H. Du, A. Kolchin and H. P. Cheng, J. Chem. Phys., 2004, 120, 1044–1054 CrossRef CAS PubMed
- S. R. Wasserman, G. M. Whitesides, I. M. Tidswell, B. M. Ocko, P. S. Pershan and J. D. Axe, J. Am. Chem. Soc., 1989, 111, 5852–5861 CrossRef CAS
- I. M. Tidswell, T. A. Rabedeau, P. S. Pershan, S. D. Kosowsky, J. P. Folkers and G. M. Whitesides, J. Chem. Phys., 1991, 95, 2854–2861 CrossRef CAS
- K. G. Grabar, P. C. Smith, M. D. Musick, J. A. Davis, D. G. Walter, M. A. Jackson, A. P. Guthrie and M. J. Natan, J. Am. Chem. Soc., 1996, 118, 1148–1153 CrossRef CAS
- J.-Y. Tseng, M.-H. Lin and L.-K. Chau, Colloids Surf., A, 2001, 182, 239–245 CrossRef CAS
- A. Yoshikawa, M. S. Gordon, V. F. Sidorkin and V. A. Pestunovich, Organometallics, 2001, 20, 927–931 CrossRef CAS
- Y. Li, J. Huang, R. T. McIver and J. C. Hemminger, J. Am. Chem. Soc., 1992, 114, 2428–2432 CrossRef CAS
- M. J. Tarlov and J. G. Newman, Langmuir, 1992, 8, 1398–1405 CrossRef CAS
- M. H. Schoenfisch and J. E. Pemberton, J. Am. Chem. Soc., 1998, 120, 4502–4513 CrossRef CAS
- M. M. Kreevoy, E. T. Harper, R. E. Duvall, H. S. Wilgus and L. T. Ditsch, J. Am. Chem. Soc., 1960, 82, 4899–4902 CrossRef CAS
- H. Rieley, G. K. Kendall, F. W. Zemicael, T. L. Smith and S. Yang, Langmuir, 1998, 14, 5147–5153 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2014 |