
Study of different-sized sulfur-free expandable graphite on morphology and properties of water-blown semi-rigid polyurethane foams
Wei Luo,
Yi Li,
Huawei Zou* and
Mei Liang*
The State Key Laboratory of Polymer Materials Engineering, Polymer Research Institute of Sichuan University, Chengdu 610065, People's Republic of China. E-mail: hwzou@163.com; liangmeiww@163.com
First published on 31st July 2014
Abstract
In this paper, EG with different nominal particle sizes (70 μm, 430 μm and 960 μm) and different loadings from 0 to 50 pphp were considered to investigate the effect of expandable graphite on the morphology and properties of water-blown semi-rigid polyurethane foams (SPFs). Experimental results showed that a higher content of EG in the SPF was more conducive to the formation of a barrier layer after expansion and exhibited better flame retardancy. Horizontal burning test confirmed the conclusion that the EG-430 μm and EG-960 μm with various loadings could effectively enhance the flame retardant properties of the SPFs. However, EG-70 μm could not improve the fire behavior of the composite due to a weak ability to form an efficient char layer on the surface of the SPF. Thermo gravimetric analysis (TGA) indicated that EG had a positive effect on the thermal stability of SPFs. In addition to the flame retardant behavior, effects of EG on the density, pore structure, thermal properties, mechanical properties and damping properties of the SPFs were also investigated.
1. Introduction
Serving as one of the most important commercial products of polyurethanes, semi-rigid polyurethane foam (SPF) is a material with very interesting properties such as low density, high specific strength and good energy absorption capacity, and has now been widely used as an interior automobile material, industrial cushioning material and packaging material.1–4 However, SPF is a highly flammable material due to its porous, open-cell structure, low degree of crosslinking and low density, which limits its further use in many fields.5 Incorporation of flame-retardant additives into foams is the most popular approach to improve the flame retardancy of polyurethane foams (PUFs). Recently, a large range of flame retardants such as halogen, phosphorus, and nitrogen based compounds has been used in PUFs.6 Due to the highly toxic and potentially carcinogenic brominated furans and dioxins formed during combustion, halogen based compounds, as a high efficiency fire retardant additive, is currently restricted in SPFs. A great deal of attention has been paid to halogen-free fire retardant additives.7Expandable graphite (EG), as a halogen-free flame retardant, is prepared by the intercalation of a variety of intercalation agents8,9 and can be rapidly exfoliated, to some extent, hundreds of times over its initial volume when subjected to high temperature, and forming a low density, ‘worm-like’ morphology (Fig. 2c). The ‘worm-like’ structure forms a low-density, thermal insulating layer on the heating surface which prevents the polymer from further degrading. Because of outstanding effect, EG has been widely used as flame retardant additives in polymers.10–12 Numbers of studies were reported about EG modified polyurethane foams (PUFs).13–20 Unfortunately, these studies are mainly focused on the rigid polyurethane foam, whereas the effect of EG on modified semi-rigid or flexible polyurethane foams has been few reported. Moreover, great majority of EG are made from sulfuric acid and will release large amounts of SO2 and SO3 during its exfoliation process and has corrosive sulfur residual, which limited its applications in nuclear power plants, electronics, aerospace and automobile industries.21
Flame retardant properties of the EG/PUF composites depend strongly on the EG size, foam density and the EG content.22–24 In this paper, three large-span particle sizes of sulfur-free EG (70 μm, 430 μm, 960 μm) were filled into water-blown SPFs with different content as a flame retardant additives. The flame retardant properties and thermal stability of the composites were evaluated by LOI, Underwriters Laboratories 94 (UL-94)34 standard and thermogravimetric analysis. Dynamic mechanical analysis (DMA) was employed to examine the damping properties of the composites. Moreover, the effects of EG flakes on the viscosity of the reaction mixture, density, morphology and mechanical properties of the foam were also discussed.
2. Experimental
2.1 Raw materials
Polymethylene polyphenyl isocyanate (PAPI) was supplied by Badische Anilin-und-Soda-Fabrik (BASF) Co. The –NCO content by weight and average functionality are 30.5 and 2.7 respectively. Polyether polyol 3050A was purchased from GaoQiao Petro. Co. (Shanghai, China). According to the manufacturer, typical hydroxyl number is 55.8 mg KOH per g with a functionality of 3. Low molecular weight polyethylene glycol, used as a chain extender, was synthesized in our laboratory with a functionality of 2. Dabaco DC5188 was used as a surfactant. Catalysts Dabaco 2040 and stannous octoate, employed for the formation of urethane and cyclotrimerization of isocyanate, were purchased from Air Product (China) and Chengdu Chemical Reagent Co. (Chengdu, China). Distilled water was used as a chemical blowing agent. Sulfur-free expandable graphite, with a broad particle size distribution, was provided directly by the manufacturer with a density of 2.2 g cm−3, ash of 0.9%, moisture of 0.6%, volatile of 10%, pH value of 7.0 and initial expansion temperature at about 160 °C, the digital micrographs of three kinds of EG are shown in Fig. 1. | ||
| Fig. 1 The digital micrographs of EG with different sizes ((a), EG-70 μm; (b), EG-430 μm; and (c), EG-960 μm). | ||
Materials used in this work classed into two components as component A and component B. Component A is from a mixture of substances containing polyether polyol, chain extender, surfactants, catalyst, blowing agents, and EG. Component B is isocyanate (PAPI).
2.2 Foam preparation
Before the foam preparation process, EG was sieved into three different particle sizes by a series of grading sieves. The average nominal particle sizes in diameter of EG used in this study were 70 μm, 430 μm and 960 μm, which were statistic values based on the micrograph measurement, respectively. The expanded volumes of EG-70 μm, EG-430 μm, EG-960 μm at high temperature are showed in Fig. 2. | ||
| Fig. 2 The ‘worm-like’ structure of the expanded EG. | ||
The EG-filled SPFs were prepared through a one-shot, free-rise method. The chemical compositions used to prepare the EG/SPF composite in this study are presented in Table 1. All components except EG and PAPI were mixed and stirred together with an electric stirrer until a uniform mixture was obtained. Then measured EG was added and the mixture (component A) was stirred for 3 min. PAPI was added into the mixture in a certain weight ratio and then stirred fast for 15 s. And then the mixture was quickly poured into an cube mold with a dimension of 250 mm × 250 mm × 250 mm. Finally, the foams were aged at room temperature for 24 h before characterizations. In this work, The EG loading was from 0 to 50 pphp. Foam samples were cut into required shape for the evaluation of different properties.
| Classification | Chemicals | phpa | Weight% |
|---|---|---|---|
| a pphp = parts per hundred of polyol by weight. | |||
| Component A | Polyol 3050A | 100 | 40.98–51.55 |
| Chain extender | 6 | 2.46–3.09 | |
| Dabaco DC5188 | 2 | 0.82–1.03 | |
| Dabaco 2040 | 2 | 0.82–1.03 | |
| Stannous octoate | 0.5 | 0.20–0.25 | |
| Distilled water | 2.5 | 1.02–1.29 | |
| EG (different particle size) | 0–50 | 20.50–0.00 | |
| Component B | PAPI | 81 | 33.20–41.75 |
3. Characterization
3.1 Viscosity of the reaction mixture (component A)
The EG filled component A was analyzed in a dynamic rheometer (Bohlin Gemini 2000, Malvern, British) in the solution state. The samples were studied under a steady-state mode. All of the samples were tested in a frequency sweep range from 0.05 to 100 Hz with a strain of 1%. The samples were added in a parallel-plate with a diameter of 40 mm, and the rheological tests were performed at 25 °C with a gap distance of 1 mm.3.2 Apparent density
The apparent densities of the samples were carried out according to the standard ISO 845:2006. The values of the apparent density were calculated as a mass–volume ratio.3.3 Compression test
The compression strength and modulus of all samples were measured with a universal electronic tensile machine (Instron 5567, USA). The size of the specimen was 30 mm × 30 mm × 30 mm and the rate of compression process was set at 3 mm min−1 for each sample. The compressive stress at 25% strain in parallel to the foam-rise direction was performed according to the standard ISO 844:2004.3.4 Morphology characterization
The morphology of the foams before burning was examined through a scanning electron microscope (SEM, JSM-9600, JEOL, Japan) with accelerating voltage of 5 kV. Samples were cut into flakes with a blade and then sputter-coated with gold for SEM observations.The morphology of EG with different sizes and interfaces of burned 50 pphp EG/SPF composites were observed by a digital microscope with enhanced depth of field (VHX-1000, KEYENCE, Japan).
3.5 Burning behaviors
The burning behaviors of EG filled SPFs were evaluated by limiting oxygen index test, horizontal and vertical burning test.The limiting oxygen index (LOI) was measured by a HC-2 oxygen index test instrument (Jiangning, China) according to the standard ISO 4589-2:1996 with a sheet size of 125 mm × 10 mm × 10 mm.
The horizontal and vertical burning test was conducted according to UL-94 and ASTM-D 635 with a sheet size of 125 mm × 13 mm × 10 mm.
3.6 Thermo gravimetric analysis (TGA)
Thermogravimetric measurements were conducted with a thermogravimetric analyzer (NETZSCH TG 209F1 Iris, Germany) under dry nitrogen gas with a flow rate of 60 mL min−1. The relative mass loss of the samples was recorded from 50 °C to 650 °C with a heating rate of 10 °C min−1. The samples had an approximate weight of 4 mg.3.7 Dynamic mechanical analysis (DMA)
Dynamic mechanical analysis was carried out in a DMA Q800 (TA Instruments, USA). Samples with dimensions of 50 mm × 11 mm × 6 mm were tested using a cantilever bending model at a frequency of 5 Hz and a heating rate of 3 °C min−1 from −95 °C to 60 °C.4. Results and discussion
4.1 Viscosity of the reaction mixture (component A)
It is known that the synthetic of the polyurethane follow the equation:
In the processing and preparation of SPF by the one-shot, free-rise method, component B was added into component A and then high speed stirring about 2000 rpm was used to promote this synthesis reaction (see 2.2). In this case, the viscosity of component A plays a crucial role in the molding process.22 Excessive high viscosity of component A will cause incomplete reaction during the foaming and slow the rise of the foam. To study the effect of EG on the viscosity of the component A and eliminate its effects on the molding process of SPF, we conducted viscosity tests on the component A before the foam preparation. As shown in Fig. 3, it is clear that EG-70 μm had a greater tackify effect on the viscosity of component A than EG-430 μm and EG-960 μm. Such phenomenon could be attributed to the larger numbers of EG-70 μm particles, which obstructed the movement of molecular chains. As the EG content increased from 0 to 50 pphp, the viscosity of EG-70 μm filled component A showed a dramatic increase from 1.58 Pa s to 45.05 Pa s, while this values were only 5.48 Pa s and 3.49 Pa s for the other two samples. Moreover, viscosity of the component A decreased gradually with the increase of shear rate, this was anticipated due to the orientation of the molecular chains and graphite flakes during the test.
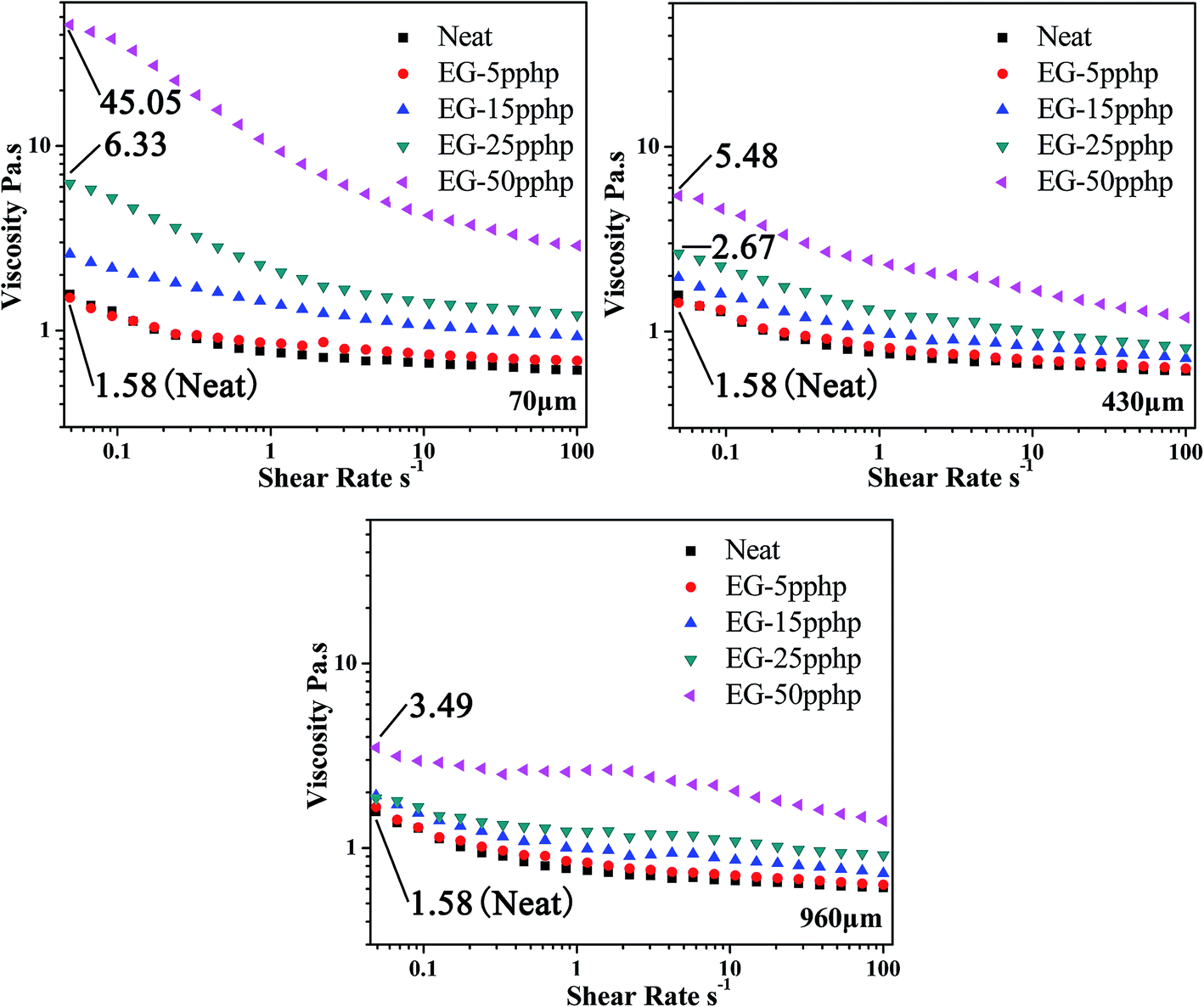 | ||
| Fig. 3 Dynamic rheological analysis results of the component A. | ||
4.2 Density and mechanical properties
Foam density is a very important parameter, which affects the mechanical properties of polyurethane foams.25 What had been demonstrated is that strength, modulus and energy absorption abilities of a material improve linearly with the increase of foam density.23 Consequently, the density of polyurethane foam materials, to a great degree, determines SPF's applications. As shown in Fig. 4, increasing the amount of EG in SPF from 5 pphp to 50 pphp resulted in a distinct increase in the apparent density of the foam, because the higher density of EG (2.2 g cm−3) than that of the neat SPF. However, three particle size of EG showed a different effect on the density of SPF. The density of the SPF filled with EG-70 μm increased from 76.42 kg m−3 for the neat foam to 116.18 kg m−3, which was greater than that of EG-430 μm per SPF and EG-960 μm per SPF composites. This was probably due to the fact that the smaller particle size of EG had a “nucleating” effect during the foam formation, and then the average cell size of the composite decreased. Similar result was reported in Bian Xiang-Cheng’s study.23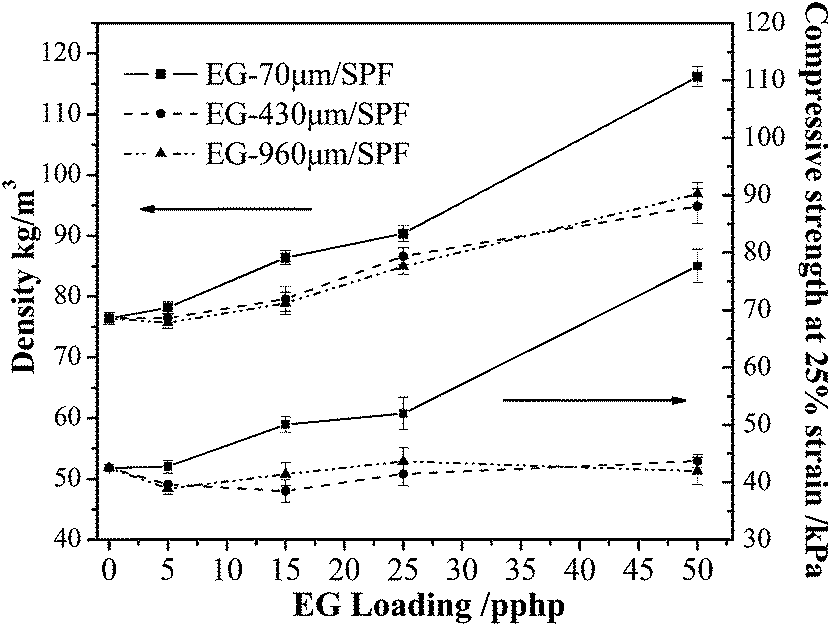 | ||
| Fig. 4 Density and compression strength of SPFs with different EG content. | ||
As a materials used in packaging industry, mechanical properties are particular important parameters in determination of the SPF's applications. However, it has been reported incorporating filler into polyurethane foam results in inferior mechanical properties, and the distribution of filler in the matrix largely determines the performance of the composites.26 In the present case, the effect of EG flake loading on the compressive strength properties of SPF was studied by the specific compressive strength (compressive strength/density) measurement. Table 2 and Fig. 4 show the specific compressive strength and compressive strength at 25% strain of the EG/SPF composite with different EG loadings. It is clear that SPFs filled with EG-70 μm demonstrated an erratic increase in compressive strength while the other two systems showed a slight decrease. The increase in compressive strength of EG-70 μm per SPF composite could be mainly due to the increase in the density. The presence of EG-70 μm in the foam matrix restricted the deformation of the foam. However, the loading of EG-430 μm and EG-960 μm distributed between the cell walls and formed many defects in composite, which attribute to the weak adhesion between the graphite flakes and the foam matrix (Fig. 5d). As a result, the compressive strength of EG-70 μm per SPF composites increased while the other two systems showed a slight decrease.
| EG content (pphp) | Density (kg m−3) | Compressive strength at 25% strain (kPa) | Specific compressive strength [kPa (kg−1 m−3)] | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 70 μm | 430 μm | 960 μm | 70 μm | 430 μm | 960 μm | 70 μm | 430 μm | 960 μm | |
| 0 | 76.42 | 42.53 | 0.557 | ||||||
| 5 | 78.19 | 76.45 | 75.65 | 42.72 | 39.64 | 38.91 | 0.546 | 0.519 | 0.514 |
| 15 | 86.42 | 79.63 | 78.92 | 50.06 | 38.52 | 41.39 | 0.579 | 0.484 | 0.524 |
| 25 | 90.37 | 86.65 | 84.96 | 51.97 | 41.47 | 43.69 | 0.575 | 0.479 | 0.514 |
| 50 | 116.18 | 94.87 | 96.89 | 77.67 | 43.70 | 41.96 | 0.669 | 0.461 | 0.433 |
 | ||
| Fig. 5 SEM micrographs of the pure foam and EG/SPF composites. ((a). Pure foam; (b). EG-70μm–50 pphp; (c). EG-430μm–50 pphp; and (d). EG-960μm–50 pphp). | ||
It was noted that the trend of apparent density and compression strength of EG/SPF composite were similar as the increase in EG loading. It suggested that changes in the mechanical strength of the foam were greatly related to the corresponding foam density.
4.3 Morphology analysis
In general, the mechanical properties of foam depend on not only the rigidity of the polymer matrix but also the cell morphology of foam. Therefore, it is of interest to observe the morphology of foam. Fig. 5a show the SEM cell micrograph of pure SPF. The open-cell structure could be found clearly in the neat SPF. Besides, and no collapse was observed in the cell system, indicating that the pure foam had a uniform cell structure.As presented in Fig. 5b, the small graphite flakes were intercalated and dispersed uniformly in the foam matrix. It is seen that the cells were not as regular as those in neat SPF, but are approximately spherical. As shown in Fig. 5(c and d), when the particle size of EG was in the same order as the cell, all the graphite flakes did not located in the struts but between the cell walls even at 50 pphp loading amount. This result was in good agreement with the study of EG-containing rigid polyurethane foam.16,22 For different particle size and loading of EG, no obvious cell bubble collapse was observed in the composites.
To better understand the distribution of the graphite in the foam, a digital microscope with enhanced depth of field was employed to observe the foam surface. As shown in Fig. 6(a and c), the increase in particle size of EG led to a decrease of graphite particle numbers in the foam (50 pphp). This decrease also indicated the uniformly distribution of EG particles. Furthermore, it could be deduced that the stirring process during the foam preparation had a damaging effect on the graphite flakes according to the micrographs (Fig. 6c).
 | ||
| Fig. 6 The morphology of EG/SPF composites. ((a), EG-70μm–50 pphp; (b), EG-430μm–50 pphp; (c), EG-960μm–50 pphp; and (d), interface between EG flake and foam matrix). | ||
Obvious gaps existed between the graphite flakes and SPF matrix as the arrow shown in Fig. 6d, indicating the poor interaction between the EG particles and the organic foam matrix.
4.4 Combustion properties
Limiting oxygen index (LOI), horizontal and vertical burning tests were employed to investigate the flame retardant behavior of EG/SPF composites. | ||
| Fig. 7 Limiting oxygen index (LOI) values of the EG/SPF composites. | ||
The flame retardant mechanism of EG/SPF composite is shown in Fig. 8. When the composite was subjected to fire, the external graphite expanded fast into a low density, ‘worm-like’ morphology, and the volume increased more than 200 times instantly. This structure, as shown in Fig. 1, embedded in the surface and formed thermal insulating layers on the heating surface of the foam. This expanded EG (also called burned layer or char layer) could hinder the oxygen diffusion, limit the heat penetration, and restrict mass transfer from SPF matrix to the heat source, thus prevent the matrix from further degrading. Similar results was reported by Modesti et al.14
 | ||
| Fig. 8 Flame retardant mechanism of EG/SPF composites. | ||
It should be emphasized that the presence of EG-70 μm lead the LOI value of the composite first decreased and then increased slowly. This may be attribute to the smaller expansion volume of EG-70 μm (Fig. 1) couldn't form thermal insulating layers and improve the flame retardancy of SPF. What's more, this low-density ineffective barrier layer (Fig. 9) is beneficial to the air exchange during the combustion and the degradation of SPF matrix promoted. The slight increase of LOI value at high EG loading was related to the increase in apparent density of the foam (see Fig. 3).
 | ||
| Fig. 9 Microscope images of the interface of burned 50 pphp EG/SPF composites. | ||
 | (1) |
| Sample | Neat | 5 pphp | 15 pphp | 25 pphp | 50 pphp |
|---|---|---|---|---|---|
| a HB-3, the burning rate is less than 40 mm min−1.b HB-2, the flame front pass the 25 mm reference mark and self-extinguishing.c HB-1, the flame front does not pass the 25 mm reference mark and quenches after the ignition source is removed. | |||||
| EG-70 μm | 72.86 mm min−1 | 72.2 mm min−1 | 70.2 mm min−1 | 86.7 mm min−1 | 73.3 mm min−1 |
| EG-430 μm | 56.6 mm min−1 | HB-3-38.7a mm min−1 | HB-2-9.2b mm | HB-1c | |
| EG-960 μm | HB-3-39.8 mm min−1 | HB-1 | HB-1 | HB-1 V-0 | |
It can be seen that the burning rate of the composites showed a different response with the content of EG increased from 0 to 50 pphp. The EG-70 μm showed almost no effect or even increased on the burning rate of SPF while the EG-430 μm and EG-960 μm had a significantly decrease. According to the data in Table 3, the EG-430 μm per SPF composite reached the horizontal burning test of HB-3 rating at a 15 pphp loading while the EG-960 μm per SPF achieved only at the loading of 5 pphp. What's more, only the EG-960 μm per SPF composite with 50 pphp EG loading could pass the vertical burning test and classified as V-0 rating because of its good fire retardant property.
The results of horizontal burning test were in good agreement with the LOI testing (Fig. 7). The data also proved that EG with larger particle size and at higher loading could efficiently improve the flame retardant properties of SPFs for practical use. The presence of EG-70 μm had no improvement, or even negative effect on the flame retardant of SPFs.
4.5 Thermal stability
Fig. 10 shows the thermogravimetric (TG) and derivative curves (DTG) behaviors of pure SPF and SPFs filled with three different particle sizes of EG under nitrogen. Both the neat and EG-containing samples took a two-step degradation process from nearly 230 °C to 400 °C.17,27 The representative thermal analysis data of the samples are listed in Table 4. The initial decomposition temperature (T5wt%) was the temperature at 5% weight loss, while the maximum weight loss temperature (Tmax) was taken from the peak value of the DTG curve. Results revealed that the presence of EG had a significant increase in T5wt% of SPF systems. Moreover, the larger size and higher content was conducive to the raise of T5wt% of the EG-containing SPFs. It is believed the exfoliated graphite flakes formed during the heat treatment process was not beneficial to the penetration of heat, improving the thermal stability of SPFs. Larger particle size of EG and high content in the SPFs have a positive effect on the conformation of the isolation layer which showed higher thermal stability. The DTG thermograms show that SPFs had a slight effect on the value of Tmax while in the presence of EG. In addition, in the case of SPF/EG, a shoulder peak was observed between 260 °C and 320 °C. This may be due to the oxidation and degradation of the polymer matrix promoted by residual nitric acid exists in the EG interlayer.27 | ||
| Fig. 10 The TG–DTG curves of the neat and EG filled SPFs. | ||
| Sample | Neat | EG-70 μm | EG-430 μm | EG-960 μm | |||
|---|---|---|---|---|---|---|---|
| 25 pphp | 50 pphp | 25 pphp | 50 pphp | 25 pphp | 50 pphp | ||
| T5wt% (°C) | 247.0 | 269.0 | 271.4 | 269.8 | 270.5 | 275.4 | 279.6 |
| Tmax (°C) | 369.6 | 371.9 | 372.6 | 366.9 | 365.7 | 368.4 | 367.8 |
| Residue in 650 °C (%) | 11.10 | 27.6 | 35.02 | 29.23 | 36.23 | 27.95 | 32.21 |
As shown in Table 4, the residue of EG/SPF composites primarily relied on the content of EG. The EG filled SPF exhibited higher residues than the neat foam at high temperature. Moreover, the greater the loading of EG was, the higher the weight of residual will be obtained.
4.6 Dynamic mechanical analysis
As SPF is used as package materials, the damping performance of the SPF composites is of great significance.28 Dynamic mechanical analysis (DMA) is a widely used technique to examine the viscoelastic features of PU foams.29,30 It reveals some key parameters of polymer materials (e.g., the glass transition temperature (Tg), the energy dissipation and the stiffness). Here, the effect of EG particles on the damping properties of the SPFs was measured. The change in loss factor (tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ) of pure SPF and EG/SPF composites with temperature were presented in Fig. 11.
δ) of pure SPF and EG/SPF composites with temperature were presented in Fig. 11.
 | ||
| Fig. 11 Dynamic mechanical properties of EG/SPF composites. | ||
Comparing the peak area of the tanδ curve is an effective way of evaluating the damping properties of composite. Larger peak area indicates better damping property. Table 5 show the peak areas of tanδ curves of EG/SPF composites. It could be found that the addition of the EG leading slight increase on the damping properties. This increase was attributed to not only the increasing energy loss caused by friction between the interfaces of EG and matrix, but also the energy dissipation effect of pore volume when the foams suffer deformation. As the EG content increased, the existed of large numbers of EG particles restricted the deformation of the foam, which resulting in the decrease of peak area. Meanwhile, due to relatively small cell size, the energy dissipation effect of pores in composite filled with EG-70 μm was not significant. The minimum peak area (12.920) was observed for the composite of 50 pphp EG-70 μm content, as presented in Table 5. Moreover, due to the larger surface area and cell size, the EG-960 μm per SPF composites had a better damping property compare to the other two systems.
δ curve obtained from DMA analysis
| Contents | Peak area | ||||
|---|---|---|---|---|---|
| Pure foam | 5 pphp | 15 pphp | 25 pphp | 50 pphp | |
| EG-70 μm per SPF | 13.865 | 13.917 | 13.430 | 12.920 | |
| EG-430 μm per SPF | 13.512 | 14.701 | 13.538 | 14.430 | 14.098 |
| EG-960 μm per SPF | 14.656 | 14.444 | 14.571 | 14.490 | |
Obtaining the glass transition temperature from the tanδ curves is the most common technique.31,32 The glass transition temperature (Tg) of the pure foam and the EG/SPF composite, which indicated the peak of tanδ, are listed in Table 6. It could be found that addition of the EG reduced the Tg of the composite and the higher the content of EG the lower the value of Tg could be observed.
| Contents | Glass transition temperature (Tg) | ||||
|---|---|---|---|---|---|
| 0 | 5 | 15 | 25 | 50 | |
| EG-70 μm per SPF | −7.79 | −7.83 | −10.78 | −11.2 | |
| EG-430 μm per SPF | −5.9 | −9.82 | −8.87 | −9.65 | −11.79 |
| EG-960 μm per SPF | −8.88 | −8.49 | −6.6 | −10.12 | |
The decrease in Tg of the composite may be correlated to free volume.33 As we know, EG (especially for the large particle size of EG) have large ratio between surface area and mass. This large specific surface area in the particle–matrix interface created additional free volume which provides more space to the large-scale polymer segments for their easy movements. With the increase of the EG loading from 0 to 50 pphp, the free volumes became bigger which results in the further decrease of Tg.
5. Conclusions
The morphology and properties of semi-rigid polyurethane foam (SPF) filled with three large-span particle sizes (EG-70 μm, EG-430 μm and EG-960 μm) of sulfur-free expandable graphite were investigated.With the introduction of middle size (EG-430 μm) and large size (EG-960 μm) EG, the composites presented significant improvement in flame retardant. SPF filled with 50 pphp EG-960 μm showed the excellent fire behavior with LOI value of 31 and vertical burning test of V-0 rating. EG-70 μm, however, showed negligible or even negative effect on flame retardant of SPFs.
Thermogravimetric analysis (TGA) showed that EG could significantly improve the thermal stability of SPFs, especially for EG with the larger size and higher content. The loading of EG leaded to a slight increase in damping properties and a decrease in glass transition temperature (Tg) for each sample.
Acknowledgements
The authors would like to thank National Natural Science Foundation of China (51273118), Provincial Science and Technology Pillar Program of Sichuan (2013FZ0006), for financial support, and the Analytical and Testing Center of Sichuan University for providing SEM measurements.References
- P. Cinelli, I. Anguillesi and A. Lazzeri, Eur. Polym. J., 2013, 49, 1174 CrossRef CAS PubMed.
- M. A. Mosiewicki, G. A. Dell'arciprete, M. I. Aranguren and N. E. Marcovich, J. Compos. Mater., 2009, 43, 3057 CrossRef CAS PubMed.
- D. Q. Chen, J. Y. Yang and G. H. Chen, Composites, Part A, 2010, 41, 1636 CrossRef PubMed.
- S. Mitsuru, I. Atsushi, and M. Masayoshi, U.S. Patent, 6306, 918 Bl ( 2001).
- J. Q. Wang and W. K. Chow, J. Appl. Polym. Sci., 2005, 97, 366 CrossRef CAS PubMed.
- S. V. Levchik and E. D. Weil, Polym. Int., 2004, 53, 1585 CrossRef CAS PubMed.
- G. Camino, L. Costa and M. P. Luda, Makromol. Chem., Macromol. Symp., 1993, 74, 71 CrossRef CAS PubMed.
- M. S. Dresselhaus and G. Dresselhaus, Adv. Phys., 1981, 30, 139 CrossRef CAS.
- J. Li, L. Feng and Z. Jia, Mater. Lett., 2006, 60, 746 CrossRef CAS PubMed.
- R. Xie and B. Qu, J. Appl. Polym. Sci., 2001, 80, 1190 CrossRef CAS PubMed.
- Z. Z. Li and B. J. Qu, Polym. Degrad. Stab., 2003, 81, 401 CrossRef CAS.
- C. F. Kuan, W. H. Yen, C. H. Chen, S. M. Yuen, H. C. Kuan and C. L. Chiang, Polym. Degrad. Stab., 2008, 93, 1357 CrossRef CAS PubMed.
- M. Modesti and A. Lorenzetti, Polym. Degrad. Stab., 2002, 78, 167 CrossRef CAS.
- M. Modesti and A. Lorenzetti, Eur. Polym. J., 2003, 39, 263 CrossRef CAS.
- X. G. Zhang, L. L. Ge, W. Q. Zhang, J. H. Tang, L. Ye and Z. M. Li, J. Appl. Polym. Sci., 2011, 122, 932 CrossRef CAS PubMed.
- M. Modesti, A. Lorenzetti, F. Simioni and G. Camino, Polym. Degrad. Stab., 2002, 77, 195 CrossRef CAS.
- A. Wolska, M. Goździkiewicz and J. Ryszkowska, J. Mater. Sci., 2012, 47, 5627 CrossRef CAS.
- A. Wolska, M. Goździkiewicz and J. Ryszkowska, J. Mater. Sci., 2012, 47, 5693 CrossRef CAS.
- S. Duquesne, R. Delobel, M. L. Bras and G. Camino, Polym. Degrad. Stab., 2002, 77, 333 CrossRef CAS.
- L. Shi, Z. M. Li, M. B. Yang, B. Yin, Q. M. Zhou, C. R. Tian and J. Wang, Polym.–Plast. Technol. Eng., 2005, 44, 1323 CrossRef CAS.
- F. Kang, Y. Leng and T. Y. Zhang, Carbon, 1997, 35, 1089 CrossRef CAS.
- M. Thirumal, D. Khastgir, N. K. Singha, B. S. Manjunath and Y. P. Naik, J. Appl. Polym. Sci., 2008, 110, 2586 CrossRef CAS PubMed.
- L. Shi, B. H. Xie, J. H. Wang, C. R. Tian and M. Y. Yang, Polym. Int., 2006, 55, 862 CrossRef CAS PubMed.
- X. C. Bian, J. H. Tang, Z. M. Li, Z. Y. Lu and A. Lu, J. Appl. Polym. Sci., 2007, 104, 3347 CrossRef CAS PubMed.
- M. Thirumal, D. Khastgir, N. K. Singha, B. S. Manjunath and Y. P. Naik, J. Appl. Polym. Sci., 2008, 108, 1810 CrossRef CAS PubMed.
- M. Thirumal, D. Khastgir, N. K. Singha, B. S. Manjunath and Y. P. Naik, Cell. Polym., 2007, 26, 245 CAS.
- S. Duquesne, M. L. Bras, S. Bourbigot, R. Delobel, G. Camino, B. Eling, C. Lindsay and T. Roels, Polym. Degrad. Stab., 2001, 74, 493 CrossRef CAS.
- J. Lefebvre, B. Bastin, M. Le Bras, S. Duquesne, R. Paleja and R. Delobel, Polym. Degrad. Stab., 2005, 88, 28 CrossRef CAS PubMed.
- H. Ron, H. Kathy and A. Randy, Flexible polyurethane foams,Dow Chemical Co., 2nd edn, ( 1997) Search PubMed.
- B. D. Kaushiva and G. L. Wilkes, J. Appl. Polym. Sci., 2000, 77, 202 CrossRef CAS.
- A. Leszczynska and K. Pielichowski, J. Therm. Anal. Calorim., 2008, 93, 677 CrossRef CAS.
- J. L. Rivera-Armenta, Th. Heinze and A. M. Mendoza-Martínez, Eur. Polym. J., 2004, 40, 2803 CrossRef CAS PubMed.
- S. Jana and W. H. Zhong, Mater. Sci. Eng., A, 2009, 525, 138 CrossRef PubMed.
- UL-94: Test for Flammability of Plastic Materials for Parts in Devices and Appliances, Underwriters Laboratories Inc. (UL) (2001).
| This journal is © The Royal Society of Chemistry 2014 |