
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Kinetics of the AlCl3 catalyzed xylan hydrolysis during Methanosolv pulping of beech wood†
Martin
Schwiderski
 *a,
Andrea
Kruse
ab,
Robert
Grandl
a and
Dennis
Dockendorf
a
*a,
Andrea
Kruse
ab,
Robert
Grandl
a and
Dennis
Dockendorf
a
aKarlsruhe Institute of Technology, Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany. E-mail: martin.schwiderski@kit.edu; Fax: +49 721 608 22244; Tel: +49 721 608 26511
bUniversity of Hohenheim, Garbenstrasse 9, 70599 Stuttgart, Germany. E-mail: Andrea_Kruse@uni-hohenheim.de; Fax: +49 711 459 24702; Tel: +49 711 459 24700
First published on 8th September 2014
Abstract
In this work the kinetics of the AlCl3 catalyzed xylan hydrolysis during Methanosolv pulping of beech wood is investigated in the range of 150 to 170 °C. Herein the focus lies on the maximization of the furfural yield. Therefore the kinetic rate constants of the xylan hydrolysis, the degradation of monomeric sugars to furfural and byproducts and the degradation of furfural are presented. They are compared with literature data. The yield of furfural is limited to about 45% (mol/mol). At lower temperatures the degradation of monomeric sugars to byproducts and at higher temperatures the degradation of furfural is favoured. The catalyst concentration has almost no influence on the selectivity.
Introduction
Furfural is a high value added product with an increasing global production. Its existence as a platform chemical is justified because of its many applications. Furfural itself can be used as an extractant, fungicide or nematocide.1 Furfural can be hydrogenated to MTHF 2-methyltetrahydrofuran. MTHF can act as a biofuel component and as an alternative solvent to tetrahydrofuran.2,3 The most important application of furfural is the production of furfuryl alcohol. It can be further converted to furan resins. One of the advantages of furan resins compared to phenol/formaldehyde resins is that there is no toxic formaldehyde released. There are many other applications of furfural. A good overview is given by Zeitsch.1Furfural is produced by hydrolysis of biomass using Brønsted acids, especially sulfuric acid. The oldest furfural production technology is the Quaker oats process patented in 1923.4 Herein the reaction takes place at about 150 °C for 5 hours. The yield of furfural in this process is about 40% till 50% of the theoretical value. In addition to the low yield there is another disadvantage: except the energetic conversion there is no use of the side products cellulose and lignin in this process. The kinetics of the Brønsted acid catalyzed xylan hydrolysis to furfural is well known. The formation of byproducts is caused by a reaction of an intermediate with furfural.1 Because of this knowledge other furfural producing processes are developed. The BIOFINE and the STAKE process are operating at higher temperatures and shorter reaction times giving a furfural yield of about 70% (mol/mol).5,6 The SUPRAYIELD process exploits that furfural can't react to byproducts if it's in the vapor phase during the reaction. Another way to suppress the formation of byproducts is the in situ extraction of the formed furfural.7 Weingarten et al. report that a furfural yield of 85% (mol/mol) can be achieved while reacting xylose in a biphasic water/methylisobutylketon-system.8
Another idea to improve the furfural yield is to change the catalyst. There are some publications using Lewis acids as catalysts.9–11 Yang et al. reported that the conversion of poplar wood in a biphasic water/tetrahydrofuran medium catalyzed by alumina chloride giving a furfural yield of 51% (mol/mol). Under the same reaction conditions using hydrochloric acid the yield is 29% (mol/mol).10 Mao et al. investigated the hydrolysis of corncob with concentrated sea water.11 They observed a furfural yield of 73% (mol/mol) when a mixture of iron(III)–chloride and acetic acid is used as the catalyst system. Unfortunately, there is no kinetics of the Lewis acid catalyzed xylan hydrolysis published.
In a previous work, the ability of AlCl3 to catalyze the hydrolysis of xylan during the Ethanosolv pulping of beech wood was demonstrated.12 It is shown that in this procedure furfural is formed up to 50% (mol/mol). This work is continued in this paper. Here, the kinetics of the xylan hydrolysis during Methanosolv pulping of beech wood is investigated.
Materials and methods
Materials
All chemicals were purchased from sigma Aldrich with p.a. quality. The starting material was beech wood chips with the dimensions of about 200 mm × 100 mm × 100 mm. The chemical composition of the wood chips is given in Table 1. Experimental details are given in the ESI.† The main part of the C5-sugars is xylose. Therefore, xylan is assumed to consist only of anhydroxylose-units.| Component | Content | Component | Content |
|---|---|---|---|
| Anhydroglucose | 35.8% | Anhydrorhamnose | 0.3% |
| Anhydroxylose | 17.7% | Klason-lignin | 22.3% |
| Anhydrogalactose | 1.5% | Acid soluble lignin | 2.1% |
| Anhydromannose | 1.4% | Extractives | 1.5% |
| Anhydroarabinose | 0.6% | Others | 16.8% |
Organosolv pulping in microscale and workup
The microscale experiments were done to determine the kinetic rate constants of the xylan hydrolysis. Before the use of the wood chips they were chopped with a granulator. These chips passed a sieve with 2 mm mesh size. About 1 g of the air dried wood chips (5% moisture) were placed in a stainless steel autoclave with a volume of 10 ml. First of all, the solvents for the reactive extractions were prepared. Therefore, 133 mg, 266 mg and 532 mg AlCl3 were dissolved in 50 ml MilliQ water and filled up to 100 ml with methanol. The resulting concentration of AlCl3 is 0.01 M, 0.02 M and 0.04 M, respectively. The water–methanol ratio is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v). Then 5.6 ml of the solvent was added into the reactor. The solvent includes water and methanol, therefore a type of Organosolv process was applied. The reactor was placed in a GC-oven and heated for different heating periods. When the oven reached the desired temperature, the time was set to be zero. Therefore, only the time at this temperature is called “reaction time” here. The reactions were carried out from 150 °C till 170 °C.
:1 (v/v). Then 5.6 ml of the solvent was added into the reactor. The solvent includes water and methanol, therefore a type of Organosolv process was applied. The reactor was placed in a GC-oven and heated for different heating periods. When the oven reached the desired temperature, the time was set to be zero. Therefore, only the time at this temperature is called “reaction time” here. The reactions were carried out from 150 °C till 170 °C.
During the reaction the autoclave was mechanically rotated with 20 rpm. After the desired reaction time the oven was cooled down and the autoclave was left in the oven. After cooling down the suspension was filtered and the residue was washed four times with 5 ml methanol and then four times with 5 ml MilliQ water. The residue was dried at 105 °C over night.
The combined filtrates were filled up to 100 ml with MilliQ water and analyzed for carbohydrates and furfuralic compounds.
Organosolv pulping in larger scale and workup
There were two experiments in an increased scale done. As the starting material for these experiments uncut wood chips were used. In the first experiment 60 g of the wood chips, in the second experiment 50 g were placed in a nonstirred 500 ml stainless steel reactor. The reactor is equipped with a thermocouple element. The solutions were prepared by dissolving 667 mg AlCl3 in 250 ml MilliQ water. The aqueous solution was filled up to 500 ml with methanol. The resulting concentration of AlCl3 is 0.01 M and the water–methanol ratio is 1:1 (v/v). In the first experiment 300 ml of the solution and in the second experiment 250 ml of the solution was added. The oven was set at the desired temperature. When the oven reached the desired temperature, the reaction time was set to be zero. After cooling down the suspension was filtered and washed twice with 100 ml methanol, twice with 100 ml MilliQ water and finally once with 100 ml methanol. An aliquot of the combined filtrates were analyzed for furfuralic compounds.
Then the solution was distilled in a round bottom flask equipped with a Vigreux column under reduced pressure. About 400 ml of the first fraction (methanol rich) was distilled at a boiling point of 33–35 °C at 100 mbar. About 50 ml of the second fraction (water rich) was distilled at a boiling point of 43–45 °C at 100 mbar.
The second fraction of the distillation was three times extracted with 50 ml dichloromethane. The organic layer was dried over magnesium sulphate, the solvent was removed and 1H-NMR-, 13C-spectra and a GC-MS-chromatogram were recorded.
The distillation residue was filtered over celite and about 20 ml were extracted five times with 20 ml of ethylacetate. The organic layer was dried over magnesium sulphate, the solvent was removed and an 1H-NMR-spectrum was recorded.
Analysis of the Organosolv products
The residue was hydrolyzed using the method of Saeman.13 The residue was suspended in sulfuric acid (12 M) at room temperature for 45 min. Then 84 ml of MilliQ water was added and the mixture was refluxed for 3.5 h. After cooling down the suspension was filtered and the filtrate was adjusted to a pH of about 9 with 26 wt% ammonia solution. The resulting solution was derivatised according to Sawardeker.14 This method derivatises monosaccharide to the correspondingly alditolacetates. First 200 μl of the aqueous sample was poured into a 10 ml glass sample tube. Then 2 ml of a fresh prepared NaBH4-solution in DMSO (c = 0.5 M) was added and the solution was stirred at 60 °C for 1 h. The sample tubes were placed in a cold water bath and 100 μl of an internal standard myo-inositol, 200 μl glacial acetic acid, 400 μl 1-methyl-imidazole and 4 ml acetic anhydride were added. The reaction mixture was stirred for 10 min at room temperature. The solution was poured into a separating funnel and the sample tube was washed two times with 5 ml MilliQ water. After cooling down 4 ml of chloroform was added and the layers were separated. The organic layer was again washed twice with 10 ml MilliQ water and dried over magnesium sulfate and measured by an GC “Agilent 5890”. The column is “RT 2330”. The injection- and the FID-temperature are 275 °C and the oven temperature is 240 °C. The carrier gas is helium with a flow rate of 1 ml min−1 and the split ratio is 1:35.
An aliquot of the filtrate was measured for furfuralic compounds using HPLC on a Merck/Hitachi-device. It is a “Hypersil ODS” column. The mobile phase is a 9:1 (v/v)-mixture of acetonitrile and MilliQ water with a flow rate of 1 ml min−1. The injection temperature is 30 °C. The samples are detected with an UV-VIS-detector at a wavelength of 290 nm.
The residual solution was with 26 wt% ammonia solution adjusted to a pH of about 9 and derivatised and analyzed for carbohydrates as described above.
The NMR-spectra were recorded on 250 MHz Bruker device using CDCl3 as the solvent.
GC-MS Analysis was carried out on a Trace Ultra GC coupled with a DSQ II quadrupole MS detector from Thermo Scientific. The samples were diluted in a 1:100 ratio in dichloromethane and were analyzed using split injection at 523 K (injector temperature) on a 25 m ULTRA 2 silica column ((5% phenyl)-methylpolysiloxane) from Agilent Technologies. A constant gas flow rate of 1 ml min−1 and a split ratio of 100 with the following temperature program were applied: initial temperature 313 K, increased at 6 K min−1 to 473 K, further increased with 8 K min−1 to a final 573 K, kept for 5 min. The MS detector was operated in positive ionization mode at 70 eV with an ion source temperature of 473 K.
Post analysis method
After analyzing for the furfural concentration a sample is distilled over a Vigreux column under reduced pressure. Approximately 50 ml of the water rich fraction (boiling point ≈ 44 °C; pressure ≈ 100 mbar) is extracted three times with 50 ml dichloromethane, the organic phase is dried and 1H-NMR, 13C-NMR-spectra and a GC-MS-chromatogram are recorded.Kinetic modeling
The experimental procedure for the determination of the kinetic data is done as described above. The microscale experiments are used to determine the kinetic parameters.The determination of the kinetic parameters is done with the program Matlab.15 To calculate them the kinetic model with the BDF (backward differential formula), which is an implicit numerical method, are integrated. The difference between the measured data and the integrated kinetic model are minimized. In order to do this, this problem is treated as a least-square-problem using the Levenberg–Marquardt-formula. It is solved dependent on the kinetic parameters. The solution of the Levenberg–Marquardt-formula leads to the desired kinetic parameters.
The reaction model used is shown in Fig. 1. In 1955, Kobayashi and Sakai found that the xylan hydrolysis reaction rate decreases after about 70% conversion.16 This observation leads to a biphasic reaction model. Herein there are two types of xylan introduced, the fast and the slow reacting xylan. The amount of the fast reacting xylan is about 65% and this value differs just slightly for most of the materials.17 In this study for a catalyst concentration of 0.01 M and 0.02 M it is also calculated with an initial fast reacting xylan amount of 65%. In the case of using the 0.04 M concentrated solution the Saeman model is used.18 Herein the reaction model of Fig. 1 can be also used with an initial fast reacting xylan amount of 100%. The quantity of xylan is expressed as anhydroxylose [mol] divided by the initial anhydroxylose, 0 [mol]. Also all the other components are expressed in regard to anhydroxylose.
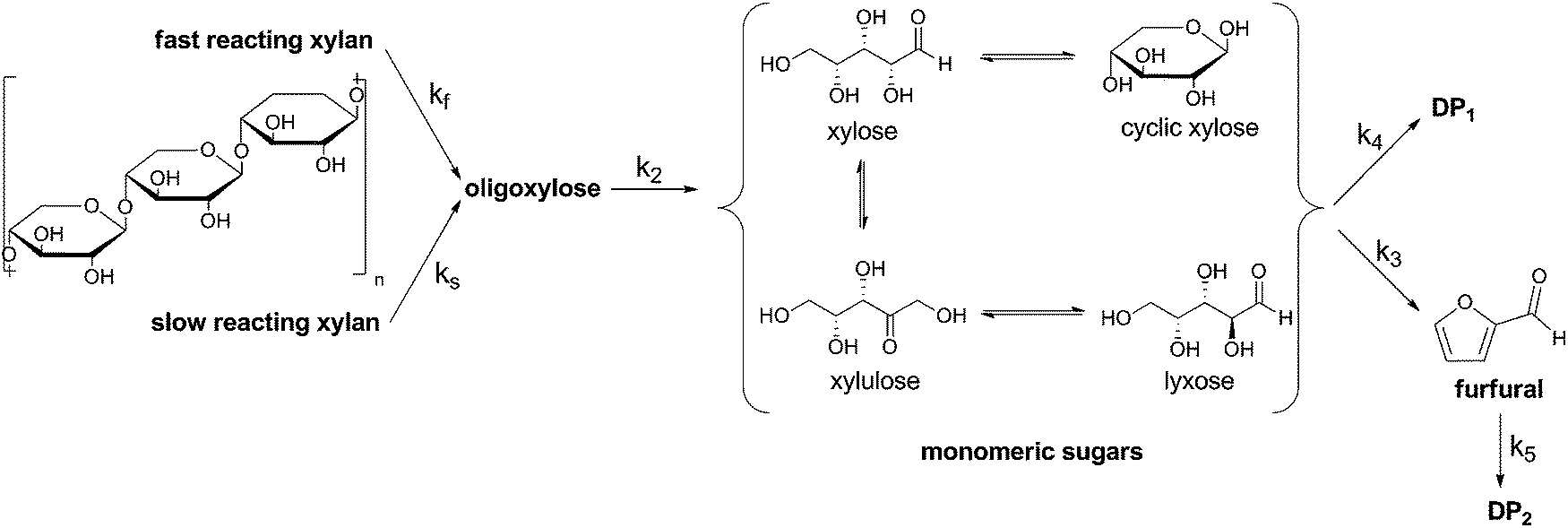 | ||
| Fig. 1 Reaction model used to determine the kinetic rate constants of the xylan hydrolysis. MS ≡ monomeric sugars; DP ≡ degradation products, in the following text also often described as byproducts. | ||
Xylose, especially if a Lewis acid is used, tend to isomerize to xylulose and lyxose.19 These carbohydrates are summed as monomeric sugars MS.
It is assumed that all the reactions follow first order kinetics. This leads to the differential rate equations (eqn (1)–(4)):
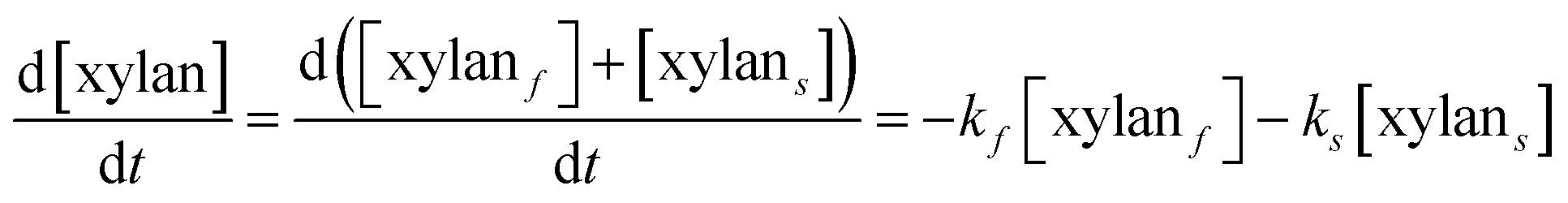 | (1) |
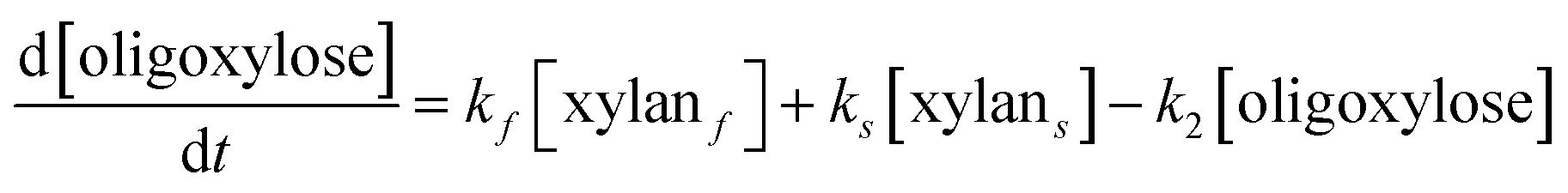 | (2) |
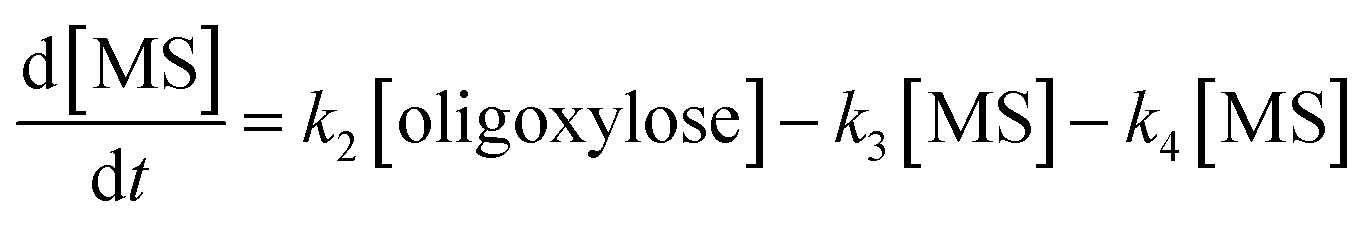 | (3) |
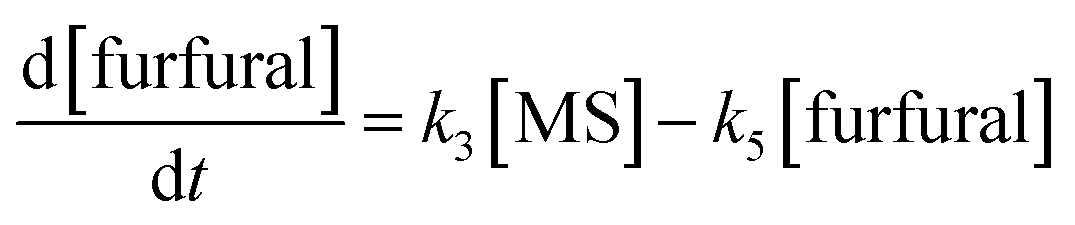 | (4) |
Results and discussion
In Fig. 2 the kinetics of the xylan hydrolysis are shown. From left to right the temperature is increasing and from up to down the catalyst concentration is increasing. For the lower concentrated (0.01 M and 0.02 M) samples A–F, the biphasic reaction model is used. For the higher concentrated samples (0.04 M) G–I, the Saeman model is used.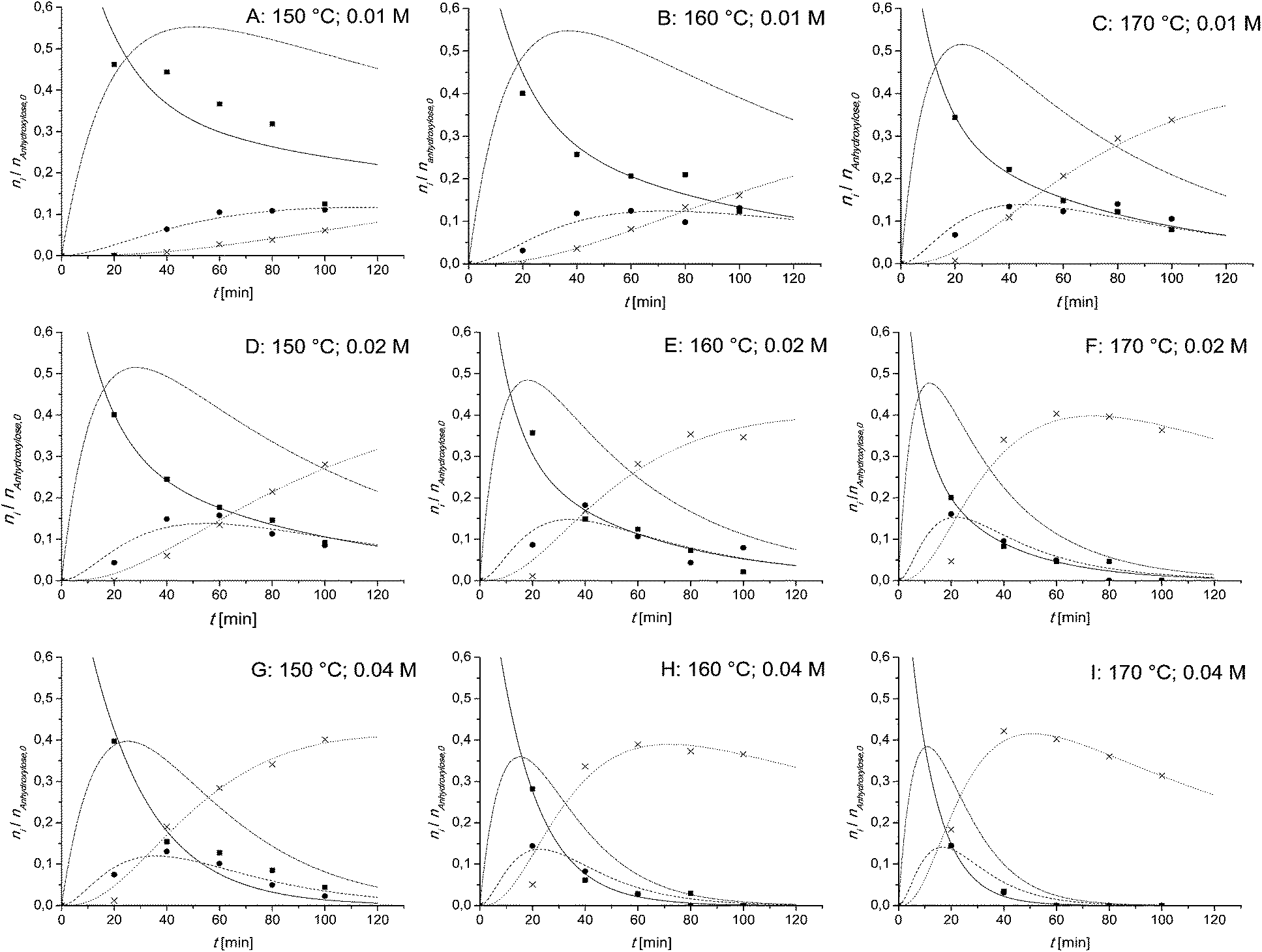 | ||
| Fig. 2 Kinetic plots of the xylan hydrolysis during Methanosolv pulping of beech wood. ■ xylan; ● monomeric carbohydrates; × furfural; solid line: fitted xylan; dashed line: fitted monomeric carbohydrates; dotted line: fitted furfural; dashed/dotted line: calculated oligoxylose; reaction conditions: (A): 150 °C, 0.01 M AlCl3; (B): 160 °C, 0.01 M AlCl3; (C): 170 °C, 0.01 M AlCl3; (D): 150 °C, 0.02 M AlCl3; (E): 160 °C, 0.02 M AlCl3; (F): 170 °C, 0.02 M AlCl3; (G): 150 °C, 0.04 M AlCl3; (H): 160 °C, 0.04 M AlCl3; and (I): 170 °C, 0.04 M AlCl3. The reaction temperature and the catalyst concentration are additionally shown on top right of each graph. | ||
As can be seen in Fig. 2 it is necessary to take the oligoxylose content into account. Its amount is in between 38 and 55% (mol/mol). It is not suitable to use the steady state approximation. The temperature has no influence of the maximum concentration of oligoxylose. Its value is constant at about 53%, 49% and 37% (mol/mol), respectively. Both parameters, the temperature and the catalyst concentration, influence the time until the oligoxylose maximum occurs. With increasing parameter values, the time that oligoxylose reaches its maximum is decreased.
The momomeric carbohydrates are in every case less concentrated. Their maximum is in between 10% and 15% (mol/mol). For low temperatures and low catalyst concentrations (A and B), the amount of monomeric carbohydrates is constant at about 10% (mol/mol) for a long period of time.
The maximum furfural amounts are given in Table 2. At 0.01 M and 0.02 M catalyst concentration the furfural yield is increasing with increasing temperature. At 0.04 M it is quite constant at 40% (mol/mol). A similar tendency can be observed, if the temperature is set as constant and the catalyst concentration varies. At 150 °C the yield increases with increasing catalyst concentration. At 160 °C and 170 °C it is quite constant. The yield of all experiments is in the range of 30 and 42% (mol/mol) comparable with the Quaker Oats process but much less in the BIOFINE process. In the BIOFINE process there is almost 70% of the theoretical furfural yield reached.
| 150 °C | 160 °C | 170 °C | |
|---|---|---|---|
| 0.01 M AlCl3 | 0.30 (717 min) | 0.36 (346 min) | 0.42 (188 min) |
| 0.02 M AlCl3 | 0.32 (258 min) | 0.39 (139 min) | 0.40 (74 min) |
| 0.04 M AlCl3 | 0.39 (72 min) | 0.42 (51 min) | 0.41 (131 min) |
The graph in Fig. 3 shows the difference between the measured and the calculated values of Fig. 2. If there were no differences between the calculated and the measured values the data points would be on the straight with the intercept through the origin and a slope of one. The mean error δ is calculated as the mean variation of a linear regression with (eqn (5)).
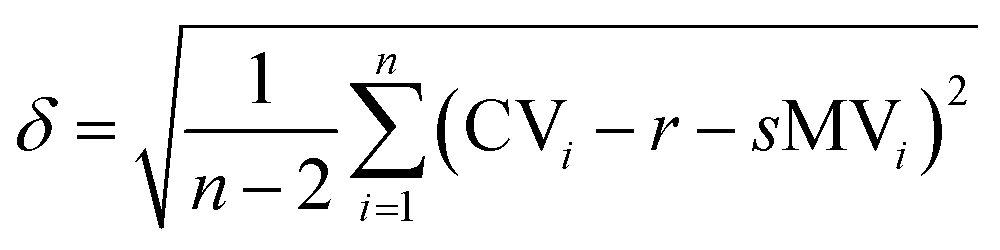 | (5) |
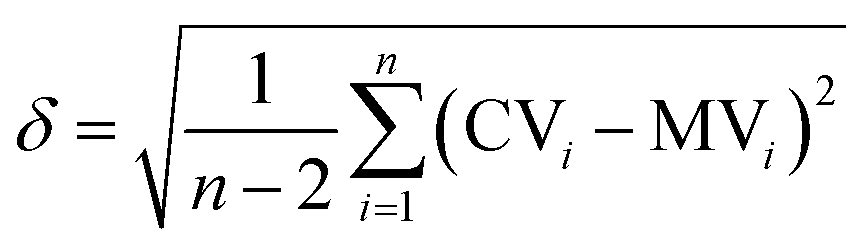 | (6) |
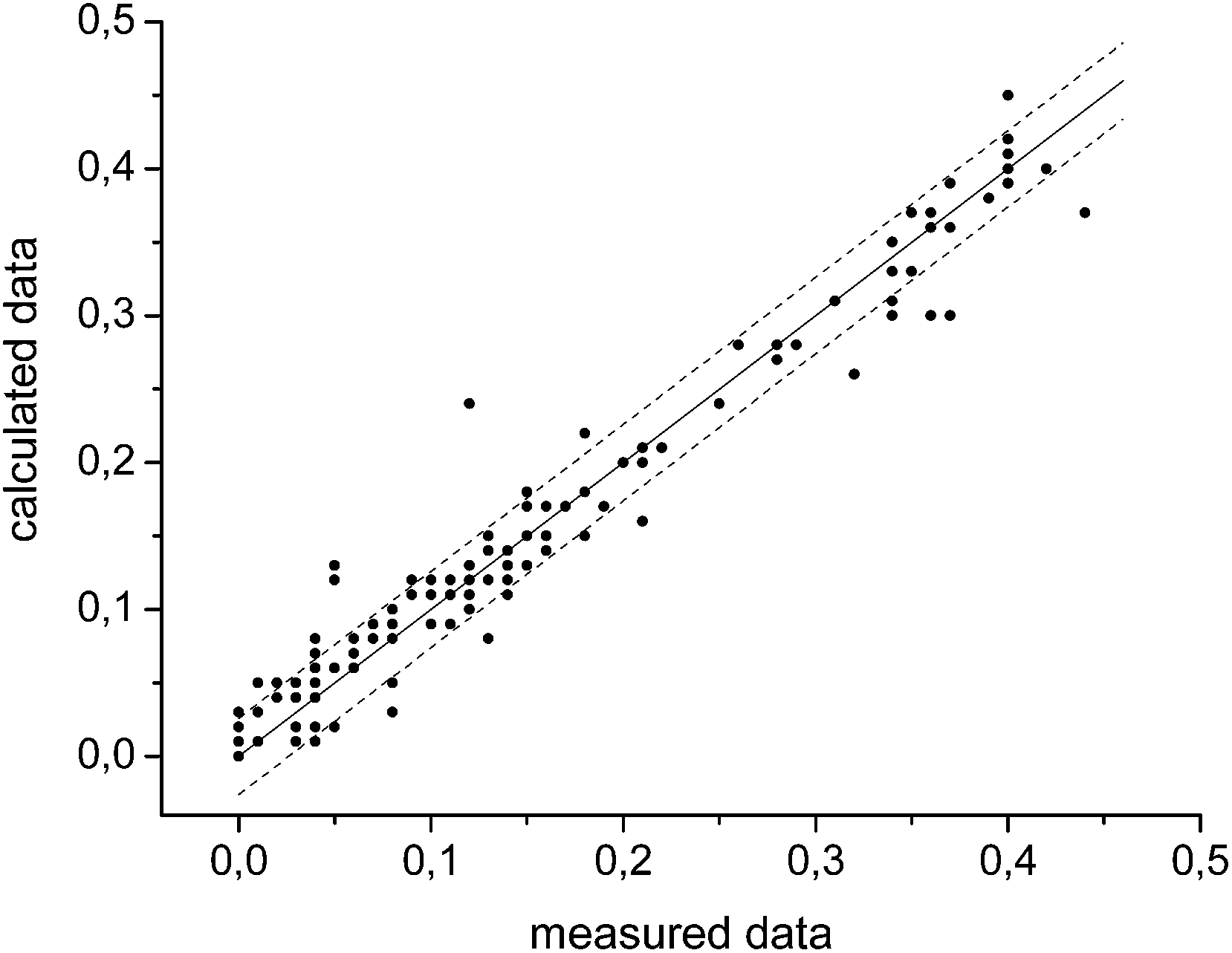 | ||
| Fig. 3 Comparison of the measured and with the Matlab calculated amounts of xylan, monomeric sugars and furfural. The solid line has an in intercept of zero and a slope of one. The dashed lines are the upper and the lower boundaries of the mean error. | ||
Temperature dependency of the kinetic constants
The reaction rate constant is dependent on the temperature and the catalyst concentration. This correlation can be expressed as the following equations (eqn (7)) and the logarithmic form (eqn (8)).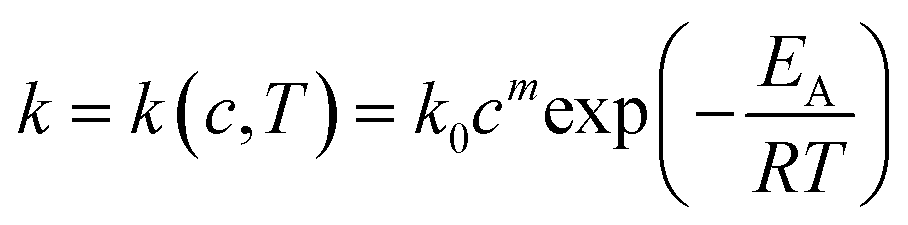 | (7) |
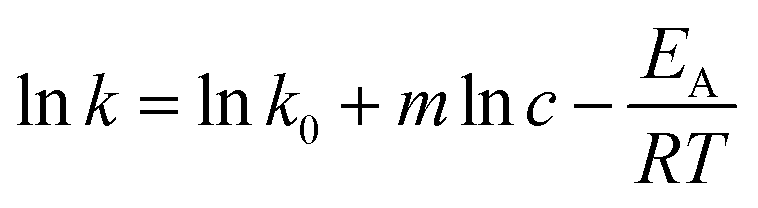 | (8) |
Bhandari et al. showed that the activation energy is independent of the catalyst concentration.20 Furthermore it is assumed that the pre-exponential factor k0 and the exponent m are temperature independent. By using (eqn (9)) the activation energy.
 | (9) |
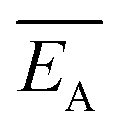 and the error
and the error 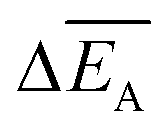 is calculated as the mean error of the mean value using (eqn (10)). In this equation j is the reaction pathway f, s, 2, 3, 4 or 5.
is calculated as the mean error of the mean value using (eqn (10)). In this equation j is the reaction pathway f, s, 2, 3, 4 or 5.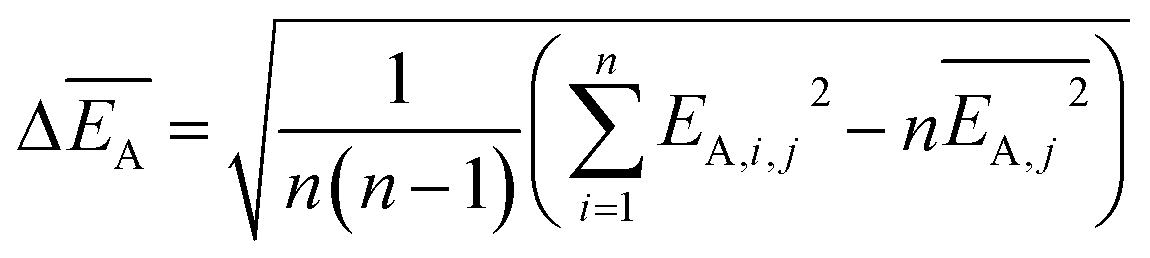 | (10) |
The mean values and mean errors of the activation energy are given in Table 3.
| k f | k s | k 2 | k 3 | k 4 | k 5 | |
|---|---|---|---|---|---|---|
| E A [kJ mol−1] | 52 ± 4 | 90 ± 5 | 83 ± 8 | 83 ± 14 | 47 ± 7 | 131 ± 9 |
| lnk0 |
15 ± 1 | 26 ± 1 | 24 ± 2 | 24 ± 4 | 14 ± 2 | 35 ± 3 |
| M | 0.65 | 1.30 | 1.26 ± 0.09 | 1.08 ± 0.12 | 0.98 ± 0.29 | 1.07 ± 0.24 |
Concentration dependency of the kinetic constants
The parameter a is determined by repeated plotting the logarithm of the rate constants against the reciprocal temperature. This time the slope is set as constant using the mean activation energies and the mean errors of Table 3. The resulting plots are shown in ESI Fig. S2.†As already mentioned before the parameter a is temperature independent but it depends on the catalyst concentration. Equate (eqn (8) and (9)), the parameter a can be expressed as (eqn (11)).
| a = a(c) = lnk0 + mlnc | (11) |
By plotting parameter a against the logarithm of the catalyst concentration the last two parameters k0 and m can be determined. The plots are shown in ESI Fig. S3.† These values are also given in Table 3. The error of m is expressed as the mean error of the linear regression of Fig. S3.† The mean error of the calculated values using (eqn (7)) is calculated using (eqn (6)). It is with 3.7% a little bit higher than the calculated mean errors with Matlab (2.6%), but it is still in a very good agreement with the experimental data.
Thus (eqn (7)) and the values from Table 3 can be used to predict the kinetics of the xylan hydrolysis in the AlCl3 catalyzed Methanosolv pulping of beech wood. Now it is examined which reaction conditions are necessary to achieve a maximum yield of furfural. Therefore the yield is shown in Fig. 4 in dependency of the catalyst concentration and the temperature. In this graph the yields are calculated with Matlab using (eqn (7)) and the parameters from Table 3. First of all it is seen that in the temperature range from 140 °C till 200 °C and in the catalyst concentration range from 0.01 M to 0.12 M the maximum furfural yield is in between 31% and 45% (mol/mol). By increasing the catalyst concentration the maximum furfural yield is also increasing. By increasing the temperature, the maximum furfural yield has a maximum in between 180 °C and 190 °C. The maximum is in the same temperature range for every catalyst concentration.
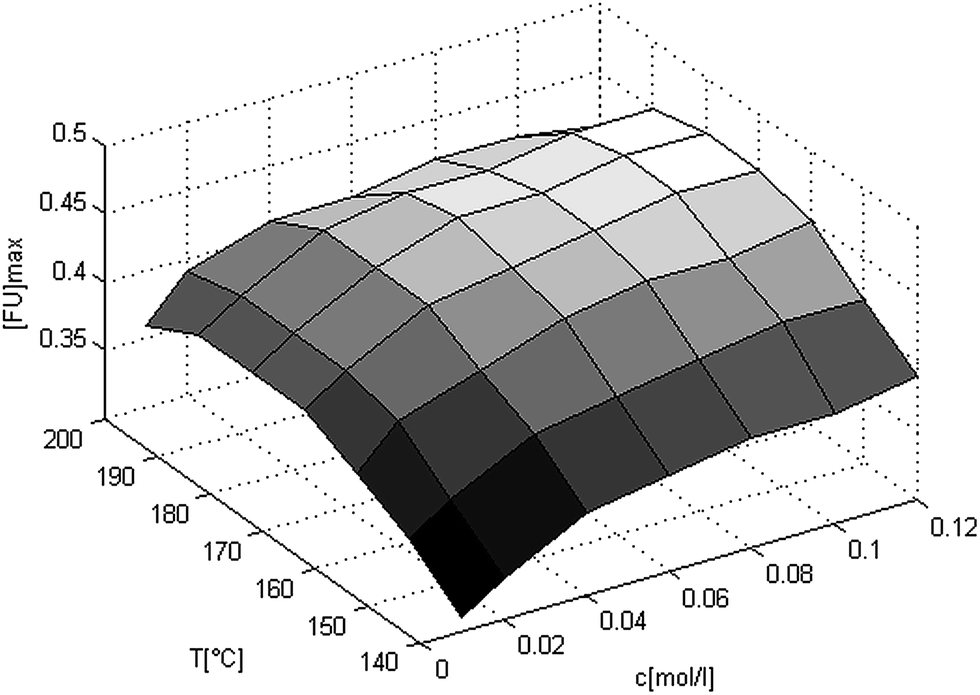 | ||
| Fig. 4 3-D-plot of the maximum furfural yields in dependency of the catalyst concentration and the temperature. The yields are calculated with Matlab using (eqn (7)) and the values from Table 3. | ||
This behavior can be explained by discussing the kinetic parameters from Table 3. Therefore the kinetic rate constants k3, k4 and k5 are plotted in dependency of the temperature and the catalyst concentration. In Fig. 5, these rate constants in dependence of the temperature at a constant catalyst concentration are shown. At lower temperatures until 180 °C, k3 has the highest value. That means until this temperature is reached, the formation of the undesired byproducts DP1 is favored. Above 180 °C, the rate constant k4 gets more dominant, thus the formation of the desired furfural is now favored. The activation energy can be discussed as the temperature dependent term of the kinetic rate constants. The activation energy of k5 shows with 131000 J mol−1 the highest value compared to the other rate constants. Thus at higher temperatures the degradation of furfural to the degradation products DP2 gets more and more dominant. Both facts, at lower temperatures k3 is more favored than k4 and at higher temperatures k5 is getting more dominant, lead to a limited yield of 30% till 45 (mol/mol) of furfural.
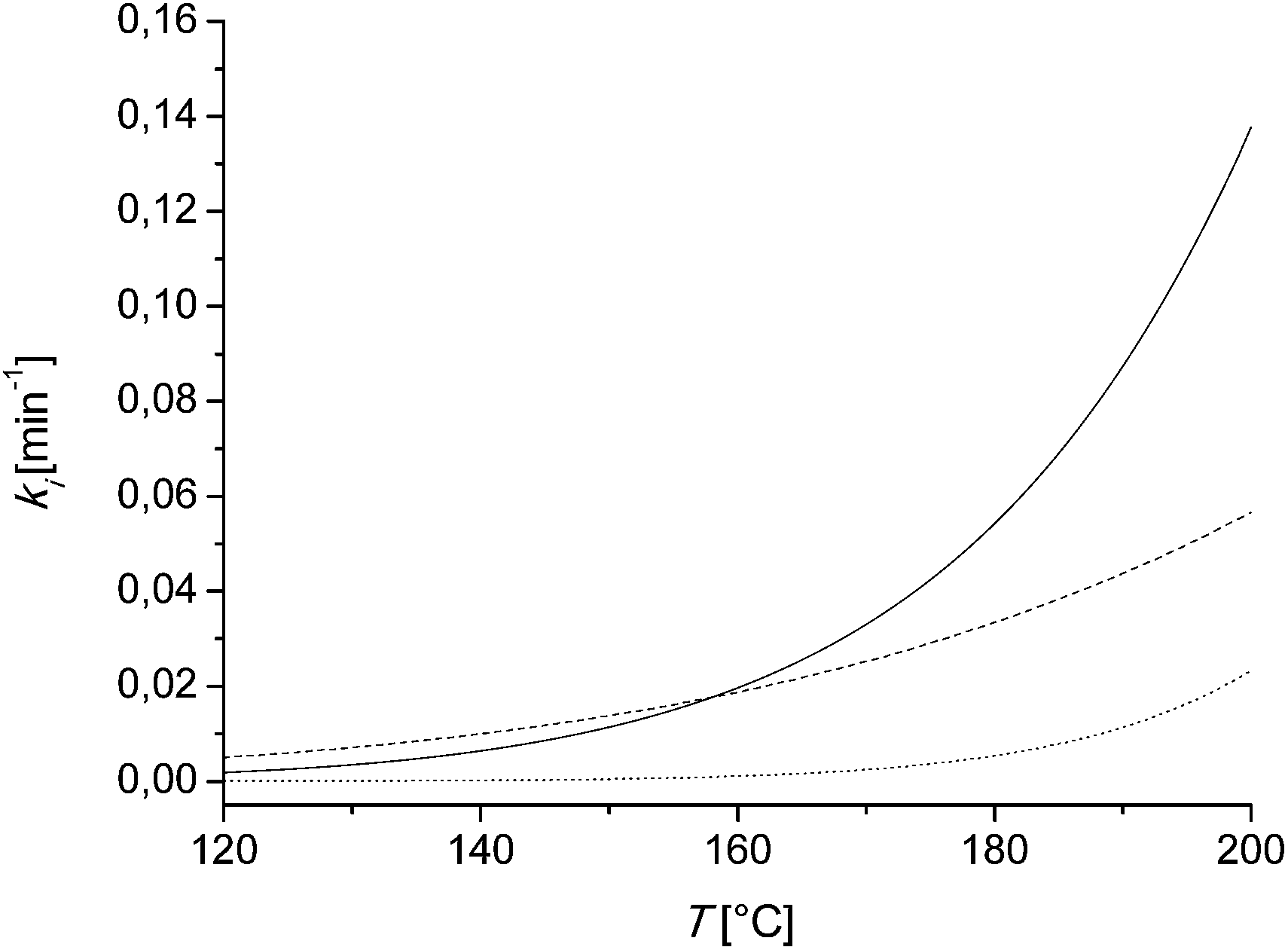 | ||
| Fig. 5 Kinetic rate constants k3 (solid line), k4 (dashed line) and k5 (dotted line) in dependency of the temperature and a constant catalyst concentration of c = 0.01 M. The rate constants are calculated using (eqn (7)) and the parameters of Table 3. | ||
The concentration dependency of the furfural yield is not such high. The parameter m is for k3, k4 and k5 quite similar in the range of one. Thus the catalyst concentration has less influence on the furfural selectivity but of course a huge influence on the reaction time (Table 2).
Results of the larger scale experiment
In this section it is checked, whether the furfural yield can be predicted using different experimental conditions and scales. Therefore, two experiments were done in a 500 ml non-stirred stainless steel autoclave. The catalyst concentration used is 0.01 M. There are several differences in between the microscale and the larger scale experiments. To determine the kinetic parameters of the xylan hydrolysis, reactors of 10 ml volume are used. The wood chips are cut with a granulator before usage. The resulting chips have a diameter of less than 2 mm. The liquor to wood ratio is constant at 5.6:1 (v/wt). During the reaction the reactor is rotated mechanically with about 20 rpm. The larger scale experiment is done in a 500 ml non-stirred reactor. The wood chips are not grinded before usage. They have dimensions of about 200 mm × 100 mm × 100 mm. The liquor to wood ratios differs with 5:1 and 6:1 just slightly from the microscale experiment. The experimental conditions are summarized in Table 4. Probably the most important difference is the heating period because of the different sizes of the reactors. In the microscale experiments it is assumed that there is no temperature difference between the reactor and the external heating source. The reactor in the larger scale experiments is equipped with a thermocouple element. The heating profiles are shown in Fig. 6. The data points are fitted as described in the following text. After filling the reactor with wood chips and the liquor the oven is tuned to 170 °C in the one and to 180 °C in the other experiment. When the oven has reached the desired temperature the reaction time is set as zero. The temperature is kept for 120 min and the oven temperature is tuned to 25 °C afterwards. It is difficult to find a function which fits the data points over the whole range. Therefore the heating period from zero till 120 min and the cooling period from 120 min till 150 min are fitted separately. A fifth grade polynom (eqn (12)) is used (Fig. 6).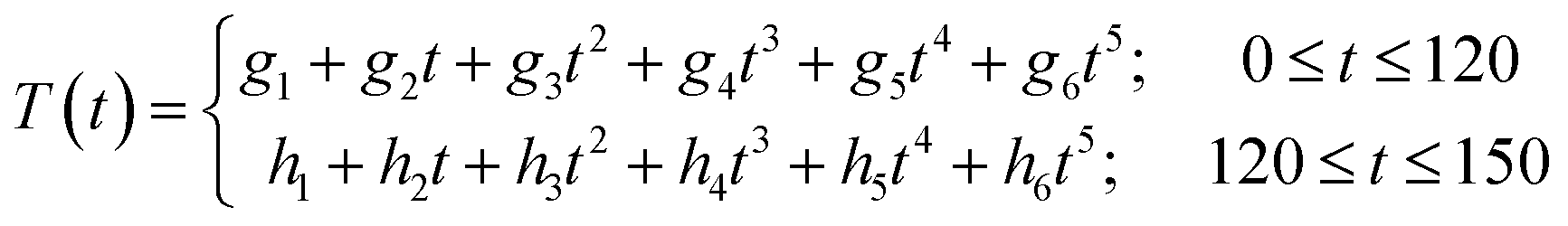 | (12) |
| Sample 1 | Sample 2 | Microscale | |
|---|---|---|---|
| Reactor volume [ml] | 500 | 500 | 10 |
| Wood chips | Uncut | Uncut | Cut |
| Cat.-conc. [mol l−1] | 0.01 | 0.01 | Various |
| Liquor:wood-ratio (v/w) |
5 | 6 | 5.6 |
| Desired oven temp. [°C] | 170 | 180 | Various |
| Calculated furfural yield | 21% | 35% | |
| Measured furfural yield | 17% | 35% |
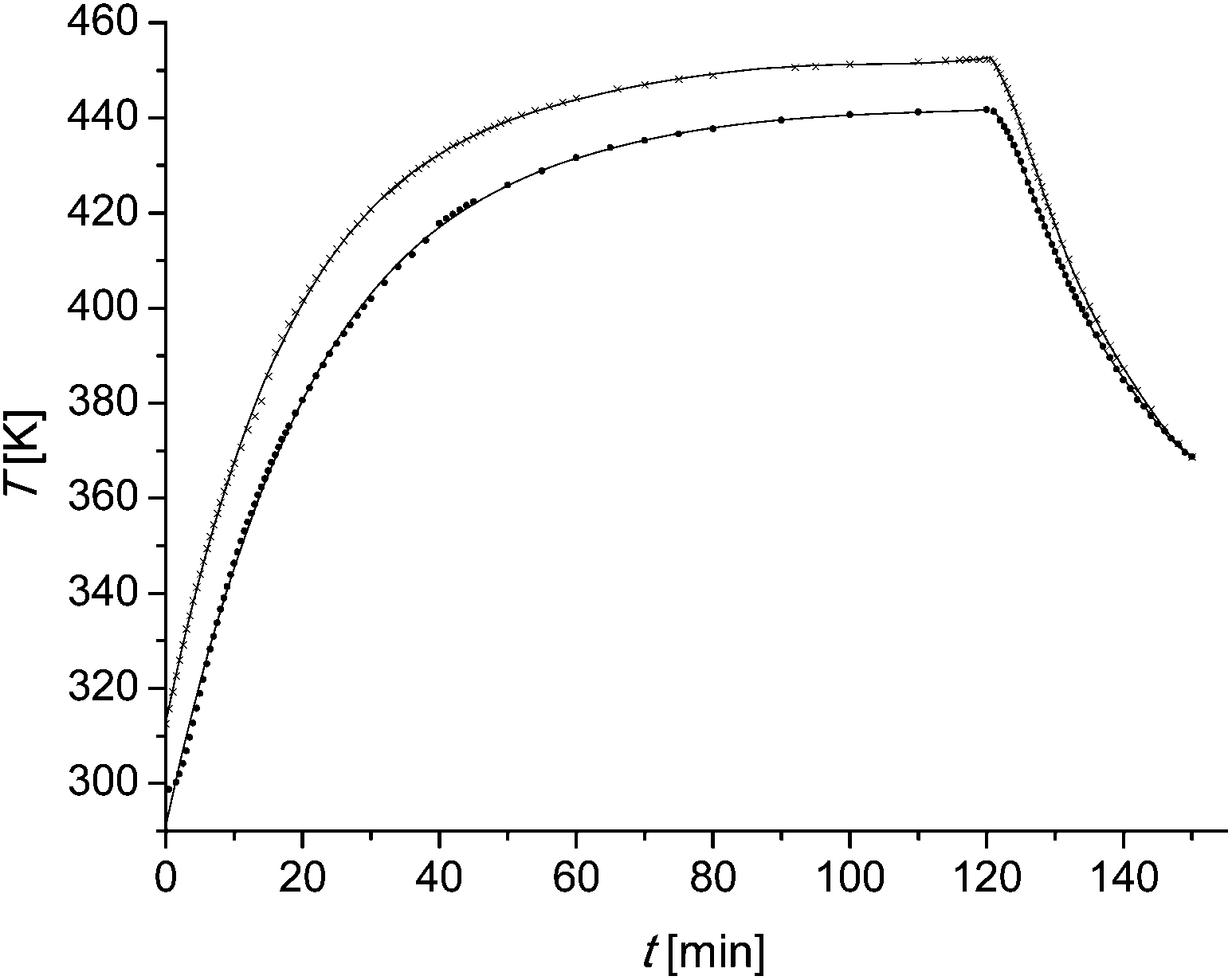 | ||
| Fig. 6 Temperature profiles of the larger scale experiments. ●: the oven temperature is set as 170 °C. ×: the oven temperature is set as 180 °C. Solid line: polynomial fits of the data points. | ||
The concentrations of xylan and its degradation products can be calculated with Matlab using (eqn (13)). This equation is equal
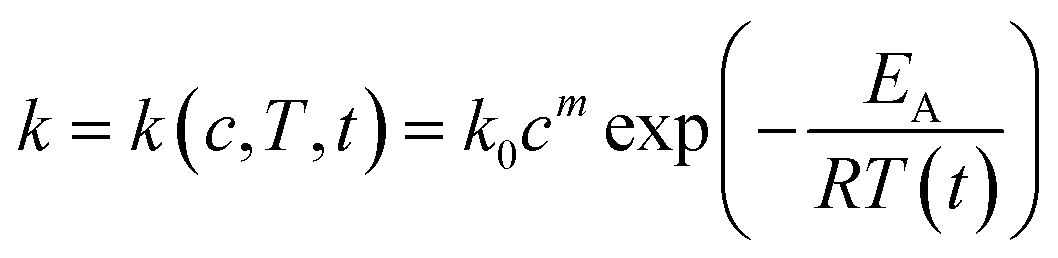 | (13) |
The calculated furfural concentration profiles are shown in Fig. 7. Up to 35 min for sample 2 and 45 min reaction time for sample 1 there are no significant changes in the furfural concentration. A comparison with the temperature profile of Fig. 6 leads to the conclusion that the reaction is very slow below 150 °C (423 K). The same can be concluded at reaction times higher than 130 min. There is neither a significant increase nor a decrease of the furfural concentration seen.
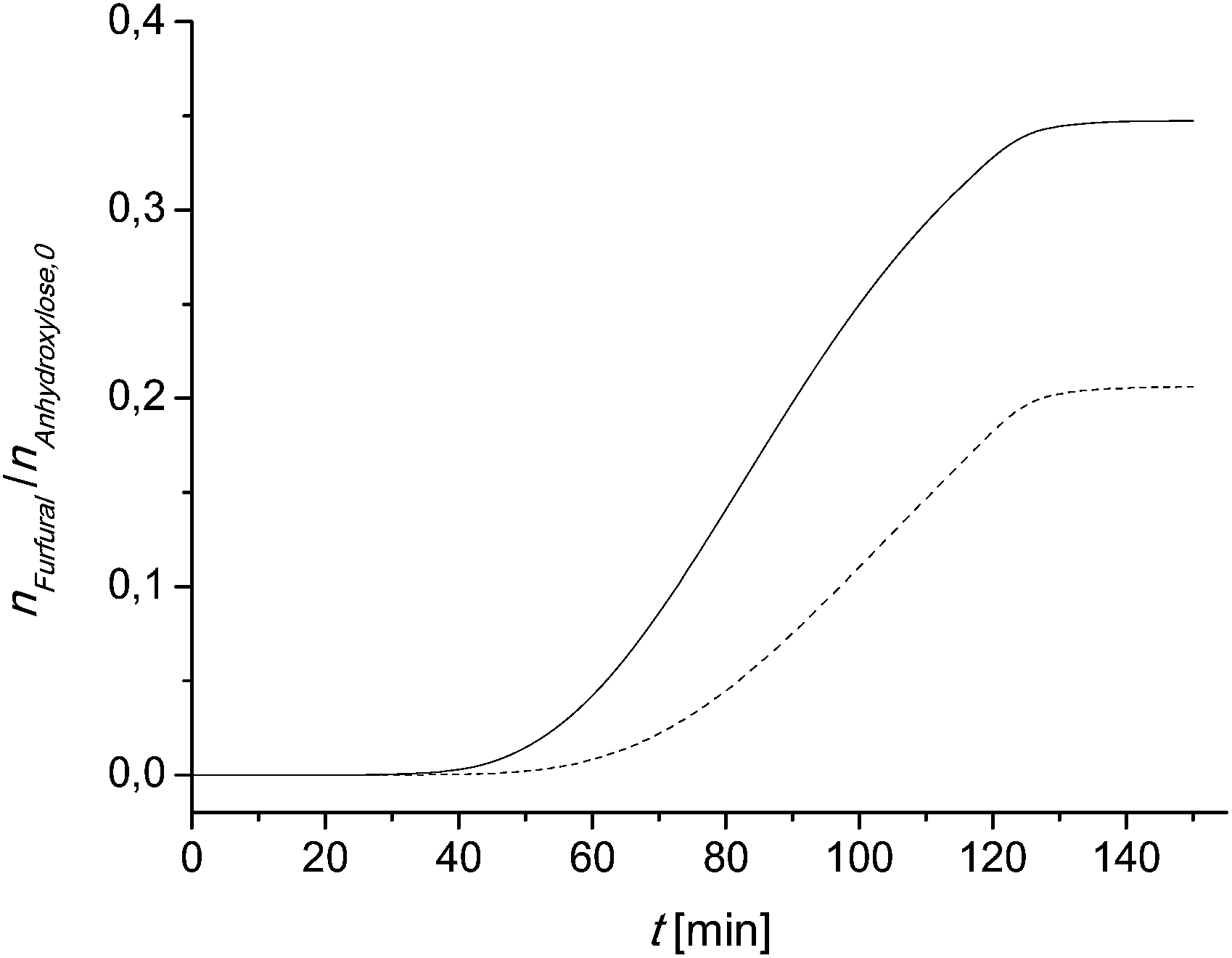 | ||
| Fig. 7 Calculated furfural concentration profile in the larger scale experiment using (eqn (7)). Dashed line: sample 1; solid line: sample 2. | ||
The calculated and the measured furfural yields are compared in Table 4. The comparison of the measured and the calculated furfural yields shows that the kinetic model, the kinetic rate equation (eqn (13)) and the parameters from Table 3 can be used to predict the furfural yield in a pretty good agreement to the experimental values.
After analyzing sample 2 for the furfural concentration, it is treated with the “Post-analysis method”. The components furfural, 5-methylfurfural and methyl lactate can be clearly identified evaluating the spectra (The spectra and the chromatogram are shown in ESI Fig. S4–S9†). It is reported that as furfural also 5-methylfurfural forms an azeotrope with water.1 The origin of the formation of 5-methylfurfural is the conversion of 5-methylpentosanes which are contained in lignocellulosic biomass. To confirm the appearance of lactic acid derivates, 20 ml of the distillation residue is extracted five times with 20 ml ethylacetate, the organic phase is dried, the solvent evaporated and an 1H-NMR-spectrum is recorded (The spectrum is shown in ESI Fig. S11†). The spectrum confirms the appearance of lactic acid or one of its derivates. M. S. Holm et al. reported that the Sn-beta-zeolite catalyzes the conversion of xylose to methyl lactate.21 C. B. Rasrendra et al. were using alumina salts for the conversion of glucose in aqueous media.22 They found that these salts catalyze not only the formation of 5-hydroxymethylfurfural but also the formation of lactic acid. Thus it can be assumed that there are two main products in the conversion of xylan in the AlCl3 catalyzed Methanosolv pulping of beech wood, furfural and lactic acid.
Comparison of the kinetic rate constants with literature data
In this work, the kinetic rate constants for the xylan degradation are investigated using AlCl3 as a Lewis acid in an Organosolv pulping process of beech wood. These data are compared with other Organosolv pulping experiments using different kinds of catalysts.Xylan hydrolysis
In Table 5 the parameters for the xylan hydrolysis under various conditions are given. The activation energies are in the case of using a Brønsted acid (entry 1) or a Brønsted base (entry 2) much higher than using a Lewis acid (entry 3) as a catalyst. Very impressive is the case of using a Lewis acid, there is much less catalyst needed to reach similar values of the parameter a.| Entry 1 | Entry 2 | Entry 3 | |
|---|---|---|---|
| Source | Spruce wood | Giant reed | Beech wood |
| Solvent | EtOH:H2O = 1:1 (v/v) |
EtOH:H2O = 2:3 (v/v) |
MeOH:H2O = 1:1 (v/v) |
| Liquor to solid (v/wt) | 6 | 6 | 5.6 |
| Catalyst | SO2 | NaOH | AlCl3 |
| Catalyst to solid (wt/wt) | 75 | 25 | 0.007 |
| E A [kJ mol−1] | 155 | frX: 74 | frX: 52 |
| srX: 141 | srX: 90 | ||
| a | 40 | frX: 21 | frX: 12 |
| srX: 35 | srX: 20 | ||
| Reference | 23 | 24 | This work |
Monomeric sugars degradation
In Table 6 the kinetic data of the degradation of xylose are shown. Entries 1–3 are using a xylose decay following first order kinetics. It is seen that either in the non-catalyzed (entry 1) or the Brønsted acid catalyzed (entry 2 and 3) reactions the activation energy is higher than in the Lewis acid catalyzed (entry 4) reaction. In a previous work it was shown that Lewis acids are able to catalyze the isomerization of xylose to xylulose.12 The degradation of the ketose xylulose is much faster than the degradation of the corresponding aldose xylose. That is why the activation energy of the Lewis acid catalyzed reaction is less compared to the Brønsted catalyzed one.Following the selectivity of the furfural formation will be discussed. Therefore the kinetic data are compared in Table 7. In the Brønsted acid catalyzed formation of furfural the byproducts are formed by reaction of the furfural and a nonvolatile intermediate of the xylose degradation.8 Thus it (a4, entry 1, Table 7) is a second order reaction. In entry 1 and entry 2 the formation of the byproducts (k4) is favored at lower temperatures. In the case of using a Brønsted acid as a catalyst this problem of the limited furfural yield at lower temperatures can be solved by distillation the reaction medium. The furfural is in the vapor phase and can't react with intermediates. In the “Suprayield” and “Batch reactive distillation process” yields in excess of 85% (mol/mol) of theoretical are reported.8,9 Conventionally furfural processes reach a yield of about 50% (mol/mol). In the Lewis acid catalyzed xylose dehydration the byproducts are not formed by reaction of furfural with an intermediate. A reactive distillation at lower temperatures would not give any yield improvements.
Furfural degradation
In Table 8 the kinetic data of the furfural degradation are shown. In the case using neither catalyst nor hydrochloric acid (entry 1–3) the activation energy is lower compared to using AlCl3 (entry 7) as a catalyst. If sulfuric acid or formic acid is used (entry 4–6) the activation energy is in the same range. This is a huge commonly difference of the concentration dependent parameter a for Brønsted and Lewis acids in entries 1 till 6. This parameter is much lower than in entry 7. This means that even at high temperatures, the degradation of furfural, if a Brønsted acid is used, is low compared to the furfural formation. Thus, using higher temperatures the furfural degradation via reaction pathway 5 (Fig. 1) can be neglected. The “STAKE” and the “BIOFINE” processes are operating at temperatures above 200 °C with a theoretical furfural yield of about 70% (mol/mol).15,16 Working at these temperatures in the AlCl3 catalyzed hydrolysis of xylan would not improve the furfural yield because of the fast furfural degradation (see also Fig. 5).Conclusions and future work
In this work the kinetics of the AlCl3 catalyzed xylan hydrolysis during Methanosolv pulping of beech wood is investigated. The xylan hydrolysis using the Lewis acid AlCl3 is faster than using a Brønsted acid or base. In addition there is much less catalyst needed for the conversion. The kinetics of the Brønsted and the Lewis acid catalyzed xylose degradation shows the same tendency: at lower temperatures the formation of byproducts and at higher temperatures the formation of furfural is favored. But the nature of the byproducts is different. In the Brønsted acid catalyzed xylose dehydration the origin of the byproducts is a reaction in between an intermediate and furfural. Using AlCl3 as a catalyst the formation of lactic acid and its derivates is observed. The amount of lactic acid is not quantified and this will be done in future work. Another important difference in between the catalysts is the degradation of furfural. In the Brønsted acid catalyzed case, this degradation can be neglected in a wide temperature range. In the Lewis acid catalyzed case, the furfural degradation plays an important role at higher temperatures and reduces the furfural yield. Thus if AlCl3 is used, a limited furfural yield of 45% (mol/mol), in xylose dehydration can be summarized: at lower temperatures the formation of byproducts (lactic acid) is favored and at higher temperatures the degradation of furfural gets more important. The selectivity for the furfural formation is nearly independent of the catalyst concentration. The xylose dehydration to furfural, the degradation to byproducts and the degradation of furfural show almost the same concentration dependencies.In this work exclusively the xylan hydrolysis is investigated. To make an assessment of the AlCl3 catalyzed Methanosolv pulping of lignocellulosic biomass there are more properties which have to be discussed. The quantity and quality of the residual cellulose is one of the key factors of an Organosolv process. Also the delignification and the properties of the Organosolv lignin play an important role of the efficiency of an Organosolv process. The cellulose and the lignin may have an influence on the kinetics of the xylan hydrolysis. However, the influence is not considered in this work.
The kinetic data of the xylan hydrolysis are obtained in the temperature range from 150 °C till 170 °C and a catalyst concentration range from 0.01 M till 0.04 M. To do a more detailed assessment of this process more experiments in a wider temperature and catalyst concentration range are necessary.
Notes and references
- K. J. Zeitsch, The chemistry and technology of furfural and its many by-products, Elsevier, 2000, vol. 13 Search PubMed.
- D. H. B. Rippin and M. Vetelino, Synlett, 2003, 2353 CrossRef PubMed.
- F. M. A. Geilen, B. Engendahl, A. Harwardt, W. Marquardt, J. Klankenmeyer and W. leitner, Angew. Chem., Int. Ed., 2010, 49, 5510 CrossRef CAS PubMed.
- H. J. Brownlee and C. S. Miner, UK Pat., 203691-A, 1923.
- S. W. Fitzpatrick, US. Pat., 4897497, 1990.
- D. B. Brown and R. Bender, US. Pat., 4211163, 1978.
- K. J. Zeitsch, US. Pat., 6743928–B1, 2000.
- R. Weingarten, J. Cho, W. C. Connor Jr and G. W. Huber, Green Chem., 2010, 12, 1423 RSC.
- J. B. Binder, J. J. Blank, A. V. Cefali and R. T. Raines, ChemSusChem, 2010, 3, 1268 CrossRef CAS PubMed.
- Y. Yang, C. Hu and M. M. Abu-Omar, Green Chem., 2012, 14, 509 RSC.
- L. Mao, N. Gao and A. Li, Green Chem., 2013, 15, 727 RSC.
- M. Schwiderski, A. Kruse, R. Grandl and D. Dockendorf, Green Chem., 2014, 16, 1569 RSC.
- J. F. Saeman, J. L. Bubl and E. E. Harris, Ind. Eng. Chem., Anal. Ed., 1945, 17, 35 CrossRef CAS.
- J. S. Sawardeker, J. H. Stoneker and A. Jeans, Anal. Chem., 1965, 37, 1602 CrossRef CAS.
- MATLAB, 2001, The Math works, Inc.
- T. Kobayashi and Y. Sakai, Bull. Agric. Chem. Soc. Jpn., 1956, 20, 1 CrossRef CAS.
- S. E. Jacobsen and C. E. Wyman, Appl. Biochem. Biotechnol., 2000, 84–86, 81 CrossRef CAS.
- J. F. Saeman, Ind. Eng. Chem., 1945, 37, 43 CrossRef CAS.
- V. Choudhary, A. B. Pinar, S. I. Sandler, D. G. Vlachos and R. F. Lobo, ACS Catal., 2011, 1, 1724 CrossRef CAS.
- N. Bhandari, D. G. Macdonald and N. N. Bakhsi, Biotechnol. Bioeng., 1984, 26, 320 CrossRef CAS PubMed.
- M. S. Holm, Y. J. Pagán-Torres, S. Saravanamurugan, A. Riisager, J. A. Dumesic and E. Tarning, Green Chem., 2012, 14, 702 RSC.
- C. B. Rasrendra, I. G. B. N. Makertihartha, S. Adisamito and A. J. Heeres, Top. Catal., 2010, 53, 1241 CrossRef CAS.
- M. Iakovlev, T. Pääkkonen and A. van Heiningen, Holzforschung, 2009, 63, 779 CrossRef CAS.
- A. A. Shatalov and H. Pereira, Carbohydr. Polym., 2005, 59, 435 CrossRef CAS PubMed.
- J. Qi and L. Xiuyang, Chin. J. Chem. Eng., 2007, 15, 666 CrossRef.
- W. Qi, S. Zhang, Q. Xu, Z. Ren and Y. Yan, Chin. J. Process Eng., 2008, 8, 1332 Search PubMed.
- I. Rose, N. Epstein and A. Watkinson, Ind. Eng. Chem. Res., 2000, 39, 843 CrossRef CAS.
- D. Root, J. Seaman, J. Harris and W. Neill, For. Prod. J., 1959, 9, 158 CAS.
- G. Marcotullio, M. A. Tavares Cordoso, W. De Jong and A. H. M. Verkooijen, Int. J. Chem. React. Eng., 2009, 7, A67 Search PubMed.
- K. Lamminpää, J. Ahola and J. Tanskanen, Ind. Eng. Chem. Res., 2012, 51, 6297 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details for the determination of the wood composition, graphs for the determination of the kinetic parameters, GC-chromatogram, MS-spectra and NMR-spectra are added. See DOI: 10.1039/c4ra05554c |
| This journal is © The Royal Society of Chemistry 2014 |