
Enforced conformational changes in the structural units of glycosaminoglycan (non-sulfated heparin-based oligosaccharides)†
Abstract
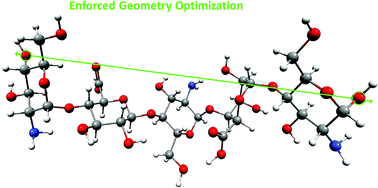
The conformational transitions in the structural units of glycosaminoglycans (GAGs) were the subject of many theoretical and experimental studies. However, relatively little is known about the mechanism of enforced structural changes that occur simultaneously in the individual components in the slightly longer fragments (oligomers) of GAGs. The complex molecular structure of mucopolysaccharides makes the correct interpretation of the AFM single molecule experiments quite complicated. Such information can be mainly obtained by theoretical methods. This paper reports the results of the Enforced Geometry Optimization (EGO) simulations of enforced structural changes in the monomeric units and selected dimeric: α-L-iduronic acid (1 → 4) α-D-glucosamine (IdoA-GlcN), β-D-glucuronic acid (1 → 4) α-D-glucosamine (GluA-GlcN), and pentameric GlcN-IdoA-GlcN-GluA-GlcN non-sulfated heparin-based oligomers. We present the theoretical analysis of the possible enforced conformational transitions.
 Please wait while we load your content...
Please wait while we load your content...

