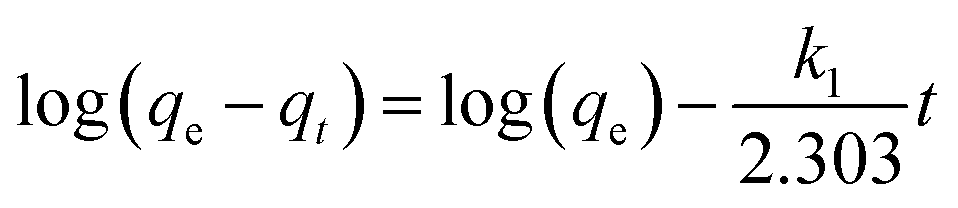DOI:
10.1039/C4RA05397D
(Paper)
RSC Adv., 2014,
4, 37600-37608
Highly efficient removal of 137Cs in seawater by potassium titanium ferrocyanide functionalized magnetic microspheres with multilayer core–shell structure
Received
6th June 2014
, Accepted 4th August 2014
First published on 4th August 2014
Abstract
In this study, a novel kind of core–shell-structured magnetic microsphere functionalized with potassium titanium ferrocyanide (KTiFC) was developed for the highly efficient removal of radioactive cesium from seawater. During the synthesis, a compact silica protective interlayer was deliberately constructed to stabilize the nano-sized magnetite cores, while preventing erosion under harsh environmental conditions. Because of high ion exchange capacity of the KTiFC functional layer, the magnetic microspheres exhibited high removal efficiency (≥97.7%) of radiocesium from 137Cs-spiked solutions (3000–35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Bq L−1) and contaminated seawater. Batch experiments revealed that adsorption equilibrium was rapidly achieved within 30 min and the maximum adsorption capacity was up to 43.09 mg g−1. Kinetic models and Langmuir/Freundlich adsorption isotherm equations were used to fit the experiment data for describing the adsorption process. Because of the favorable magnetic property, a facile separation and reclamation of the magnetic microspheres from aqueous solution was achieved under an external magnetic field. Moreover, from a practical viewpoint, the magnetic microspheres were proven to have good re-dispersion properties and long-term stability against strong HNO3 solutions (1.0 mol L−1). These magnetic microspheres are believed to hold great promise for the clean-up of radiocesium contaminated water around nuclear facilities and/or after nuclear accidents.
000 Bq L−1) and contaminated seawater. Batch experiments revealed that adsorption equilibrium was rapidly achieved within 30 min and the maximum adsorption capacity was up to 43.09 mg g−1. Kinetic models and Langmuir/Freundlich adsorption isotherm equations were used to fit the experiment data for describing the adsorption process. Because of the favorable magnetic property, a facile separation and reclamation of the magnetic microspheres from aqueous solution was achieved under an external magnetic field. Moreover, from a practical viewpoint, the magnetic microspheres were proven to have good re-dispersion properties and long-term stability against strong HNO3 solutions (1.0 mol L−1). These magnetic microspheres are believed to hold great promise for the clean-up of radiocesium contaminated water around nuclear facilities and/or after nuclear accidents.
Introduction
Substantial amounts of radioactive contamination containing harmful fission products (FPs) are generated during the operation of nuclear facilities every year. Because of their long-term threat to the ecosystem and human health, the detection and separation of these hazards has been a world-wide concern. Among the FPs, radiocesium is a major component present in radioactive liquid waste and has been involved in nuclear accidents in the past years.1 After the Fukushima Daiichi nuclear disaster in 2011, excessive levels of 137Cs (half-life 30.2 years) were detected in seawater along the coastline of the Fukushima prefecture.2 137Cs is a strong gamma emitter with high solubility, which enhances its migration through groundwater to the biosphere. Meanwhile, because of its chemical similarity to potassium, 137Cs can be easily incorporated in terrestrial and aquatic organisms.3 Therefore, recently, numerous efforts have been made to seek out effective and eco-friendly methods for the removal of radiocesium from contaminated aqueous media.4–6
Ion exchange7 is a well-established method for separating and recovering cesium from waste solutions. As a good class of inorganic ion exchanger, transition metal ferrocyanides have shown excellent adsorption ability towards cesium because of their high affinity to Cs(I) in solutions.8,9 However, the inadequate strength and unsuitable mechanical properties are major disadvantages of these sorbent materials, which render them unsuitable for industrial applications.10,11 For example, granular particles or slimes essential for liquid flow through columns are difficult to obtain by a simple precipitation reaction between soluble ferrocyanides and salts of divalent transition metals. To overcome these shortcomings, different types of support materials, such as polymers,12 biopolymers,13 colloid,11 anion exchanger resin,14 mineral oxide,15 porous silica gel particles,16 were employed for the binding/deposition of the ferrocyanides, which could then readily reduce the clogging effect and excessive pressure drop during the operation of fix-bed columns. However, the introduction of matrix materials inevitably sacrificed the operational simplicity and economic efficiency in column separation.
In recent years, magnetic nanoparticles (MNPs) have emerged as a fascinating functional material, which combine the advantages of nanomaterials with the facility of magnetic separation. Because of the unique magnetic responsivity, low cytotoxicity, and chemically-modifiable surface property,17 MNPs possess significant potential in bioseparation,18,19 environmental remediation,20 catalysis,21 enzyme immobilization,22,23 magnetic resonance imaging,24 etc. Lately, functionalization of the MNPs with transition metal ferrocyanides by deposition or coating has paved a way for the facile separation of cesium. Prussian blue and potassium nickel(II) hexacyanoferrate(II) modified MNPs have been prepared with favorable magnetism and good adsorption ability, which allowed the effective remediation of Cs-contaminated environments.13,25–27 However, from a practical viewpoint, maintaining the stability of these magnetic composites is still quite challenging. Nano-sized particles tend to form agglomerates to minimize their surface energy.28 Moreover, because the magnetic composites are mostly exposed to HNO3−-containing radioactive liquid waste for Cs adsorption, particle degradation and magnetic susceptibility reduction are unavoidable due to the dissolution and leaching of the magnetite.29 Fortunately, multilayer core–shell architecture, known as an appealing strategy for nanomaterial preparation, have offered substantial promise to solve this problem. Based upon this strategy, an impenetrable protective layer can be constructed in magnetic composites, which will not only stabilize the nano-sized particles and prevent damage to the external environment, but also provide many possibilities for further surface functionalization.30–33 However, to our knowledge, the core–shell architecture strategy has never been employed for the synthesis of ferrocyanide modified MNPs.
In present study, we report a novel kind of core–shell structured magnetic microsphere functionalized with potassium titanium ferrocyanide (KTiFC) for the effective decontamination of radiocesium. The magnetic composite exhibited high removal efficiency (≥97.7%) of radiocesium from 137Cs-spiked solutions (about 3000–35000 Bq L−1) and contaminated seawater, and could be easily separated from aqueous solution under an external magnetic field due to its favorable magnetic property. Moreover, because of the introduction of a silica interlayer, the material was endowed with good dispersion properties and excellent stability against strong HNO3 solutions (1.0 mol L−1). The structure, morphology, and magnetic properties were characterized by XRD, FT-IR, SEM and VSM. The adsorption behavior towards cesium including the adsorption kinetics and adsorption isotherm was fully investigated.
Experimental section
Chemicals
Ferric chloride hexahydrate (FeCl3·6H2O), anhydrous sodium acetate, potassium ferrocyanide (K4Fe(CN)6·3H2O), ethylene glycol (EG), tetraethyl orthosilicate (TEOS), tetrabutyl orthotitanate (TBOT), and hydroxypropyl cellulose (HPC) were purchased from Sigma-Aldrich. Cocktail Ultima Gold AB (Perkin Elmer) and 20 mL polyethylene vials (Perkin Elmer) were used for the liquid scintillation counting (LSC) measurements. All other chemicals of analytical grade, including metal nitrates, nitric acid, ammonium hydroxide and other reagents, were commercially obtained and used without further purification. Deionized water was obtained from a Milli-Q water purification system. Cesium contaminated seawater was prepared using real seawater from both the shallow and deep districts of Bohai Sea in China. A calculated amount of cesium was added to evaluate the adsorption ability of the materials.
Characterization
The Morphology of the magnetic microspheres was investigated using a LEO 1530 scanning electron microscope (SEM) with an accelerating voltage of 20 kV, coupled with energy-dispersive X-ray spectroscopy (EDX) to determine the sample composition. Powder X-ray diffraction (XRD) patterns of the products were collected on a diffractometer with Cu Kα radiation, with a scan step of 0.02° and a scan range between 10° and 80°. The Fourier transform infrared (FT-IR) spectra were recorded on a Nicolet Nexus 470 spectrometer with KBr pellets in the range 4000–400 cm−1 at room temperature. The magnetic properties (M–H curve) were measured at 300 K by cycling the external field between −10 and 10 kOe using 730T Vibrating Sample Magnetometer (VSM). The concentration of the metal ions was determined by inductively coupled plasma mass spectrometry (ICP-MS). Ultra-low background liquid scintillation counting (Quantulus 1220) from Perkin Elmer was used to measure the radioactivity.
Synthesis of Fe3O4 magnetic core
The magnetic particles Fe3O4 were prepared through a solvothermal reaction.17 Briefly, 2.70 g of FeCl3·6H2O and 7.2 g of sodium acetate were dissolved in 100 mL ethylene glycol under magnetic stirring. A homogeneous yellow solution was obtained and transferred to a Teflon-lined stainless-steel autoclave with capacity of 200 mL. The autoclave was sealed and heated at 200 °C for 8 h, followed by cooling to room temperature. The black magnetite particles were collected by magnet and washed several times with ethanol and deionized water. Finally, the product was dried in a vacuum at 60 °C for 12 h.
Synthesis of Fe3O4@SiO2 microspheres
Magnetite microspheres were coated with a thin silica layer through a sol–gel approach, forming Fe3O4@SiO2 microspheres with core–shell structure. Typically, the magnetic Fe3O4 particles (0.5 g) were treated with a 0.1 mol L−1 HCl aqueous solution by ultrasonication for 20 min. The magnetite particles were then washed with deionized water and dispersed in a mixture of absolute ethanol (200 mL), deionized water (50 mL) and concentrated ammonia aqueous solution (2.5 mL, 28 wt%), followed by the injection of tetraethyl orthosilicate (TEOS, 0.5 mL). After stirring at room temperature for 6 h, the Fe3O4@SiO2 microspheres were separated using a magnet, washed with ethanol and deionized water, and then dried in vacuum at 60 °C for 6 h.
Synthesis of Fe3O4@SiO2@KTiFC
In a typical procedure, 0.5 g Fe3O4@SiO2 particles were mixed with deionized water (3 mL), HPC (1.25 g), and absolute ethanol (400 mL) under vigorous mechanical stirring. An ethanol solution (80 mL) of TBOT (5 mL) was added dropwise followed by heating the solution to 85 °C for 150 min under reflux. The obtained product was separated with magnet, washed with ethanol several times and dried in vacuum at 60 °C for 6 h.21 The particles were then soaked in 0.5 mol L−1 K4Fe(CN)6 containing 1.5 mol L−1 HCl. After mechanical stirring for 5 h, the black particles were separated with the help of a magnet and washed with deionized water until the washing liquor was colorless. Finally, the product Fe3O4@SiO2@KTiFC was dried in vacuum at 60 °C for 10 h.
Adsorption kinetics
The adsorption kinetics were investigated by a batch operation in 10 mL plastic tube immersed in 25 °C constant temperature bath oscillator. The concentration of Cs(I) for the kinetic study was 0.3 g L−1 10 mg of Fe3O4@SiO2@KTiFC particles were adding to 4 mL of the abovementioned cesium-containing solution with HNO3 concentration of 1.0 mol L−1. The mixed liquor was shaken at about 180 rpm for different times (5–240 min). The magnetic particles were then separated using an NdFeB magnet. The aqueous solution was filtered with syringe-type filters. The concentration of the residual cesium ions in the aqueous phase was measured by ICP-MS. The adsorption capacity, qt, was used to evaluate the adsorption ability of the magnetic composite toward Cs(I). The qt (mg g−1) at time t was calculated by eqn (1):| |
 | (1) |
where C0 and Ct represent the aqueous cesium concentration at initial time and at time t, respectively; V is the volume of the solution, and M is the mass of the adsorbent.
Adsorption isotherm
The adsorption isotherm was investigated based on batch experiments. The initial cesium concentration was varied from 0.15–1.0 g L−1. The Fe3O4@SiO2@KTiFC particles were placed in contact with 4 mL of solution containing various concentration of cesium with shaking on a swing bed for 2 h. After equilibrium, the particles were separated, filtered and the residual cesium concentration was analyzed by ICP-MS.
Decontamination of radioactive 137Cs solution
Solutions containing radioactive 137Cs were prepared by diluting a stock solution to the required concentrations. 20 mg of the Fe3O4@SiO2@KTiFC particles was dispersed in 20 mL 137Cs solution in a flask. After gently shaking the flask for 90 min, solid–liquid separation was accomplished by a magnet. An aliquot of the aqueous solution was filtered through syringe-type filter and added to a vial to determine the activity by liquid scintillation counting (LSC). The measurement method has been elaborated in our previous publication.34 Here, we briefly describe it as follows. 10 mL of Cocktail Ultima AB was added to a vial for the background measurements in advance. The appropriate amount (e.g. 1 mL) of the 137Cs solutions was then added to the vial, followed by shaking to obtain a homogeneous mixture. The radioactivity of the 137Cs solutions was measured and determined by deducting the background signal.
The removal rate R (%) and decontamination factor (DF) were used to assess the adsorption performance of Fe3O4@SiO2@KTiFC particles towards 137Cs, which were defined by the following equations:
| |
 | (2) |
| |
 | (3) |
where
A0 and
Af were the aqueous cesium radioactivity in the initial solution and final solution after treatment with absorbent, respectively.
Cesium removal from seawater
5 mg of Fe3O4@SiO2@KTiFC particles were placed in contact with 4 mL of cesium contaminated seawater. After shaking for 2 h, the aqueous solution was removed and filtered through syringe-type filters. The initial and residual cesium concentration were analyzed by ICP-MS. Except for the removal rate R (%), the distribution coefficient (Kd) was defined for evaluating the Cs removal ability of the particles from seawater:| |
 | (4) |
where Ce is the equilibrium concentration of Cs(I) in the seawater.
Results and discussions
Synthesis and characterization of Fe3O4@SiO2@KTiFC
Potassium titanium ferrocyanides (KTiFC) have been recognized as the favorite inorganic ion exchanger for radiocesium clean-up. Here, by the covalent deposition of KTiFC to the silica encapsulated magnetite particles, we combined the advantages of such ion exchanger with the convenience of magnetic separation, which resulted in the facile and efficient removal of cesium in contaminated solutions. The synthesis scheme of the Fe3O4@SiO2@KTiFC core–shell structured microspheres is illustrated in Fig. 1a. First, uniform magnetite microspheres with an average diameter of approximately 300 nm were obtained via a solvothermal synthesis. The micro-morphology of the product was examined by SEM, as shown in Fig. 1b. The well-formed magnetite core particles with good mechanical strength were then utilized for surface modification and/or functionalization. Through the typical Stöber method, a silica layer with a thickness of about 20 nm was generated as a protective coating, which can be clearly observed in the TEM inset of Fig. 1c.
 |
| | Fig. 1 Schematic illustration of the fabrication procedure of Fe3O4@SiO2@KTiFC (a), and SEM study of Fe3O4 (b), Fe3O4@SiO2 microspheres (c), Fe3O4@SiO2@TiO2 microspheres (d) and Fe3O4@SiO2@KTiFC (e). The insets show the TEM image (c) and EDS spectra (d & e) of the magnetic particles. | |
As mentioned above, a large amount of acid and salts co-exist in the cesium-containing radioactive liquid waste. The introduction of a silica protective layer is crucial for preventing the erosion of the magnetite cores. Meanwhile, the silica coating also provides a friendly surface for the subsequent deposition of the KTiFC functional layer. To this end, a thin layer of amorphous TiO2 was first deposited on the Fe3O4@SiO2 microspheres by the hydrolysis of TBOT in the ethanol solution with the help of hydroxypropyl cellulose (HPC).35 The obtained Fe3O4@SiO2@TiO2 particles had a near-spherical appearance with a rough surface (Fig. 1d).
Ti signal peak was clearly observed in the EDS spectrum (Fig. 1d, inset). Finally, the Fe3O4@SiO2@TiO2 particles were immersed into a mixed solution containing potassium ferrocyanide and hydrochloric acid. Under the precipitation reaction between TiO2 and Fe(CN)64−, potassium titanium ferrocyanides were formed on the surface of the magnetic microspheres. Fig. 1e shows the SEM image of the final Fe3O4@SiO2@KTiFC particles. The presence of the Ti, K signal peaks in the EDS spectrum inset confirms the structure of the KTiFC functional layer.
The structure of the Fe3O4@SiO2@KTiFC particles were also verified by powder X-ray diffraction (XRD) and Fourier transform infrared (FT-IR) spectroscopy. Fig. 2 shows the XRD patterns of Fe3O4@SiO2 (a), Fe3O4@SiO2@TiO2 (b) and Fe3O4@SiO2@KTiFC (c). The diffraction peaks in curve (a) and (b) are in agreement with a face center cubic Fe3O4 (JCPDS card 19-629). Obviously, after coating with the SiO2 layer and TiO2 layer, the diffraction patterns of the obtained materials showed no difference compared to pure magnetite, which indicates that both silica and titanium dioxide were amorphous. The XRD pattern of Fe3O4@SiO2@KTiFC (curve c) reveals the superposition of the diffractions of a magnetite phase and a TiFe(CN)6·2H2O phase. The (200) (220) and (420) reflections of TiFe(CN)6·2H2O phase were clearly observed, which matches well with a previous study.36 Meanwhile, an evident strong resonance at 2090 cm−1 is observed in the FT-IR spectrum of Fe3O4@SiO2@KTiFC (Fig. 3d), which is the characteristic stretching vibration of cyanide groups, confirming the successful deposition of potassium titanium ferrocyanide on the surface of the Fe3O4@SiO2 microspheres. In addition, the typical adsorption bands at 582 cm−1 contributed by the stretching vibration of Fe–O band were found in all the samples. Particles coated with a silica layer exhibited a strong Si–O–Si antisymmetric stretching vibration peak at 1089 cm−1 (Fig. 3b–d). The signal of Si–O–Ti stretching vibration is around 950 cm−1, which is partially overlapped by the broad Si–O peaks.21
 |
| | Fig. 2 X-ray diffraction (XRD) patterns of Fe3O4@SiO2 (a), Fe3O4@SiO2@TiO2 (b) and Fe3O4@SiO2@KTiFC (c). | |
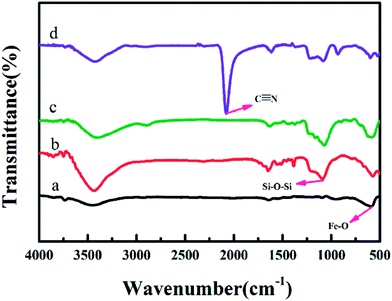 |
| | Fig. 3 FT-IR spectra of Fe3O4 particles (a), Fe3O4@SiO2 (b), Fe3O4@SiO2@TiO2 (c), and Fe3O4@SiO2@KTiFC (d). | |
Magnetic properties
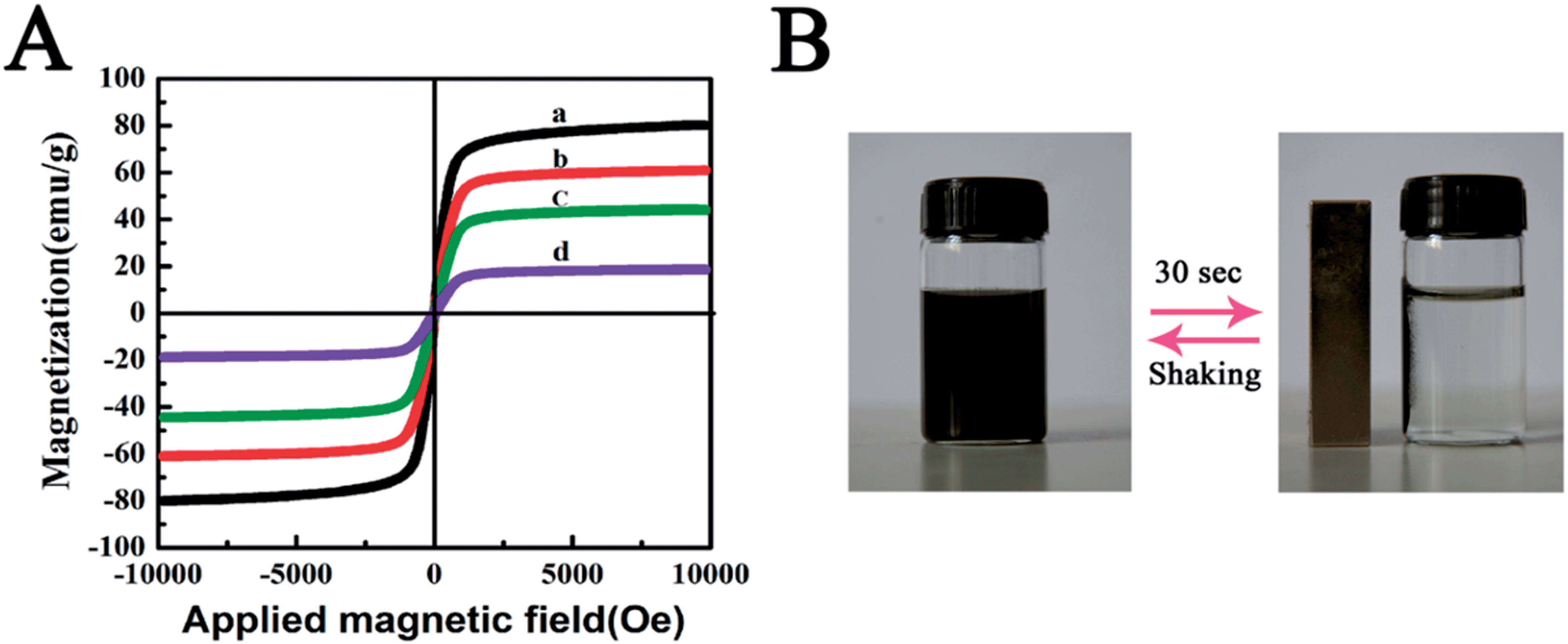
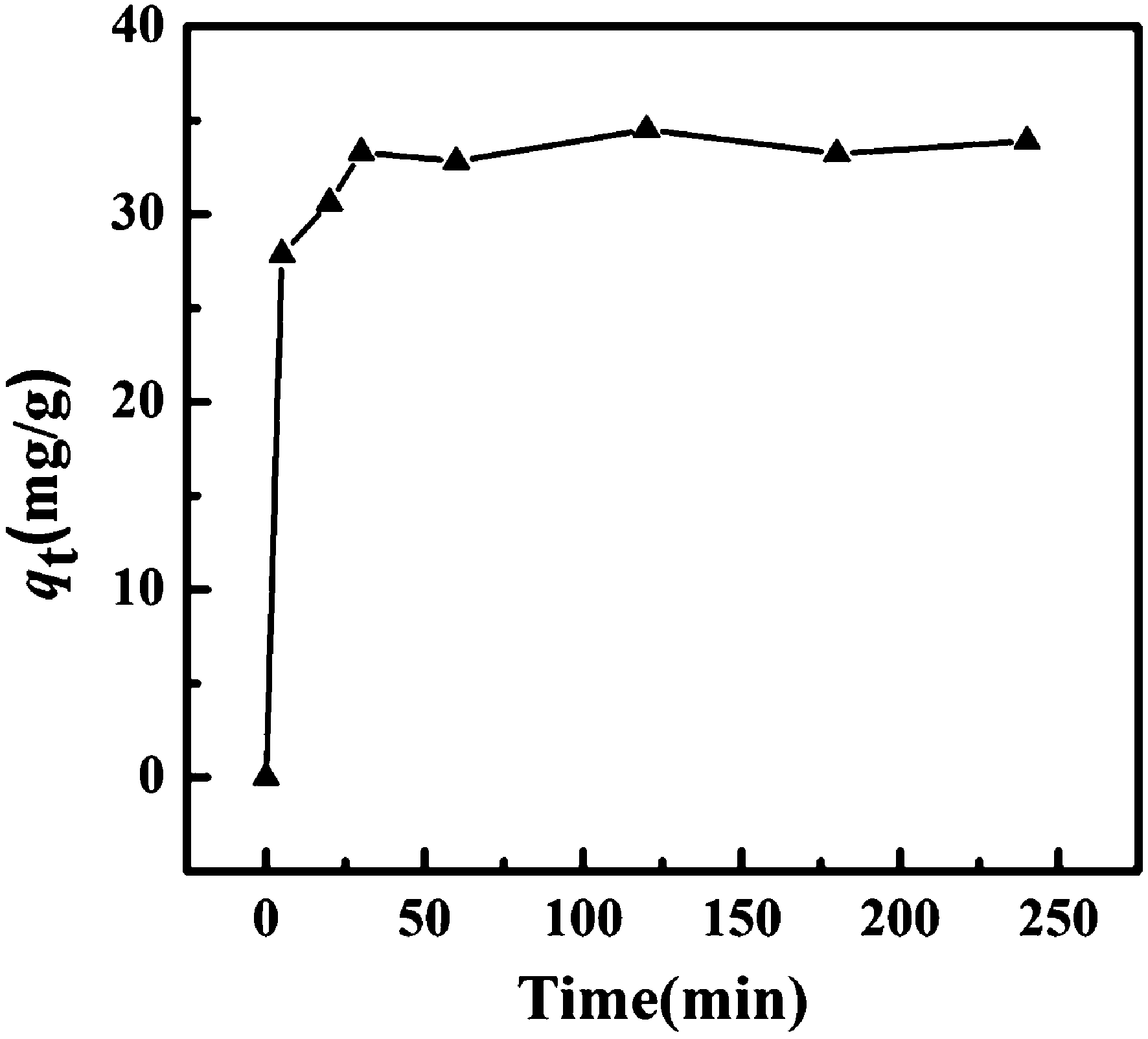
A vibrating magnetometer was used to characterize the magnetic properties at 300 K. Fig. 4A displays the magnetic hysteresis loops of Fe3O4 (a), Fe3O4@SiO2 (b), Fe3O4@SiO2@TiO2 (c), Fe3O4@SiO2@KTiFC (d). The saturation magnetization values of the magnetic composites were 80.4, 61.0, 44.1, and 18.68 emu g−1, respectively. The magnetite core and the silica encapsulated particles showed favorable magnetism, while the deposition of KTiFC resulted in a decrease in saturation magnetization. Under the applied external magnetic field, the Fe3O4@SiO2@KTiFC particles showed a sensitive response and can be quickly separated from the aqueous medium (Fig. 4B). Moreover, negligible coercivity and remanence were observed from all the hysteresis loops, which confirmed the superparamagnetism of the magnetic particles. The superparamagnetic property will facilitate the stability and redispersion of the magnetic particles in solution, which is of great significance for practical magnetic separation applications.
 |
| | Fig. 4 (A) Magnetic hysteresis loops of Fe3O4 particles (a), Fe3O4@SiO2 (b), Fe3O4@SiO2@TiO2 (c), and Fe3O4@SiO2@KTiFC (d); (B) magnetic separation–redispersion process of Fe3O4@SiO2@KTiFC. | |
Adsorption kinetics behavior
The effect of the contact time on the adsorption capacity, qt, of Fe3O4@SiO2@KTiFC towards cesium was investigated in a 1.0 mol L−1 HNO3 solution containing 0.3 g L−1 of Cs(I). After different contact time intervals between 5 min and 4 h, the mixture was separated and the aqueous cesium concentration was measured. The results are displayed in Fig. 5. The curve shows that cesium were adsorbed by KTiFC deposited magnetic materials fleetly and that the equilibrium was established after 30 min with qt ≈ 33.56 mg g−1.
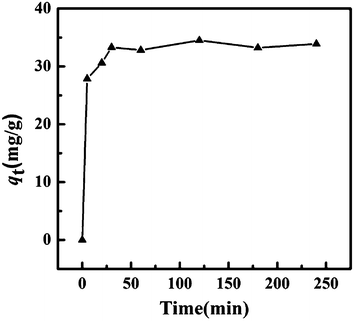 |
| | Fig. 5 Influence of contact time on the adsorption capacity of cesium. | |
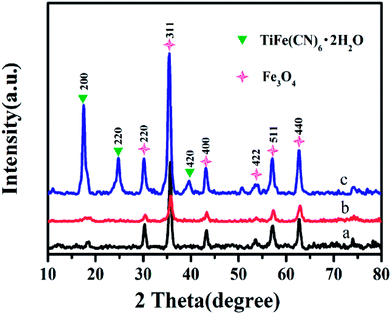
To better describe the adsorption of cesium by the Fe3O4@SiO2@KTiFC magnetic composite, two representative adsorption kinetic models, including the pseudo-first-order model37 and pseudo-second-order model,38 were utilized to fit the experimental data. The related equations are given as eqn (5) and (6):
| |
 | (5) |
| |
 | (6) |
where
qt and
qe are the adsorption capacity at equilibrium and at the time
t (mg g
−1), respectively.
k1 is the rate constant of pseudo-first-order model (min
−1) and
k2 is the rate constant of the pseudo-second-order model (g μg
−1 min
−1). Through fitting the experimental data by linear regression, the kinetic parameters
k1,
k2,
qe and the correlation coefficient
R2 were obtained and are summarized in
Table 1. Obviously, the results show that the Cs(
I) adsorption process by Fe
3O
4@SiO
2@KTiFC magnetic composite was better described by a pseudo-second-order model. The fitted curve with
R2 = 0.999 is plotted in
Fig. 6. It can be inferred that the adsorption capacity is proportional to the number of active sites on the magnetic composite, and the rate-limiting step may be chemical sorption or chemisorption involving valency forces through sharing or exchange of electrons between sorbent and sorbate.
38–40
Table 1 Kinetic parameters fitted by the pseudo-first-order model and pseudo-second-order model for cesium adsorption by the Fe3O4@SiO2@KTiFC magnetic composite
| Pseudo-first-order model |
Pseudo-second-order model |
| k1 (min−1) |
qe,1 (mg g−1) |
R2 |
k2 (g mg−1 min−1) |
qe,2 (mg g−1) |
R2 |
| 0.368 |
33.08 |
0.989 |
0.024 |
33.98 |
0.999 |
 |
| | Fig. 6 Pseudo-second-order model for the adsorption of Cs(I) by Fe3O4@SiO2@KTiFC. | |
Adsorption isotherm
The adsorption capacity of the Fe3O4@SiO2@KTiFC magnetic microspheres as a function of cesium concentration was studied in batch experiments by altering the initial concentration from 0.15 to 1.0 g L−1. The adsorption capacity, qe (mg L−1), versus concentration of cesium after equilibrium Ce (g L−1) is plotted in Fig. 7. It is observed that the adsorption capacity increased rapidly with increasing Cs(I) ion concentration when the initial concentration of Cs(I) was less than 0.3 g L−1, which may be attributed to the fact that sufficient active sites were available.41 While at a higher concentration, competition for available adsorption sites resulted in a gradual slowing of the increase in adsorption capacity.
 |
| | Fig. 7 Experimental data and the fitted curves by the Langmuir and Freundlich adsorption isotherm models. | |
The Langmuir and Freundlich adsorption isotherm models42 were applied to fit the adsorption data at equilibrium. The Langmuir model is valid for single-layer adsorption with the hypothesis that all the binding sites are free. The nonlinear form of the equation is written as:
| |
 | (7) |
where
qe and
qmax are the equilibrium adsorption capacity and the monolayer maximum adsorption capacity (mg g
−1), respectively.
K is a constant related to the affinity between the sorbent and sorbate.
In comparison, the Freundlich adsorption isotherm model is considered to be an empirical equation that is used to describe multi-layer adsorption with several kinds of adsorption sites on the surface of sorbent. The model was in the following form:
where
KF and
n are the Freundlich constants relative to the multilayer adsorption capacity and adsorption intensity, respectively.
Fig. 7 shows the experimental data and the curves fitted by the Langmuir and Freundlich models. It can be seen that the Langmuir model is more suitable for the description of the Cs(
I) adsorption by Fe
3O
4@SiO
2@KTiFC. The fitted parameters and correlation coefficient for both the models are listed in
Table 2. The correlation coefficient
R2 for the Langmuir model is up to 0.989. The maximum adsorption capacity of the Fe
3O
4@SiO
2@KTiFC magnetic microspheres toward Cs(
I) is determined to be 43.09 mg g
−1.
Table 2 Parameters of the adsorption isotherms fitted by Langmuir and Freundlich models
| Langmuir model |
Freundlich model |
| KL (L mg−1) |
qmax (mg g−1) |
R2 |
KF |
n |
R2 |
| 43.09 |
42.23 |
0.989 |
43.99 |
8.24 |
0.848 |
Selectivity
The selectivity of Fe3O4@SiO2@KTiFC toward cesium was investigated in solution containing Cs(I) and other typical interference ions such as Na(I), Ni(II), Fe(III), Sr(II), Mo(VI), Zr(IV), Ba(II), and Nd(III). The solution was prepared to simulate a real acidic radioactive liquid waste with different initial concentrations for each ion.43 The contact time was 2 h, and the phase ratio was 0.02 g mL−1. After the adsorption, the residual metal ions in the solution were measured. The distribution coefficient (Kd) was calculated and are summarized in Table 3. Obviously, the Fe3O4@SiO2@KTiFC particles showed selective adsorption toward Cs(I) in the simulated radioactive liquid waste with the highest Kd (7849.8 mL g−1). In comparison, the Kd values for all the interference ions were less than 10 mL g−1. It is worth noting that the concentration of K(I) showed a significant increase after treatment with Fe3O4@SiO2@KTiFC. This suggested that the ion exchange mechanism between K(I) and Cs(I) was responsible for the adsorption ability of potassium titanium ferrocyanide.8 Cs(I) replaced K(I) in the crystalline structures of cyanometallates, while the latter was released into the solution. Due to the size mismatch and charge unbalance, most of the interference ions could not be inserted into the crystalline structure.9
Table 3 Selectivity of Fe3O4@SiO2@KTiFC toward Cs(I)
| Ions |
Concentration (g L−1) |
Kd (mL g−1) |
| Initial |
Treatment |
| Cs+ |
0.405 |
0.003 |
7849.8 |
| Ni2+ |
0.281 |
0.254 |
5.5 |
| Fe3+ |
0.240 |
0.239 |
0.1 |
| Sr2+ |
0.157 |
0.157 |
0.0 |
| Mo6+ |
1.805 |
1.717 |
2.6 |
| Zr4+ |
0.129 |
0.124 |
2.0 |
| Ba2+ |
0.405 |
0.379 |
3.4 |
| Nd4+ |
2.170 |
2.104 |
1.6 |
| Na+ |
1.138 |
1.112 |
2.5 |
| K+ |
0.002 |
1.457 |
— |
Radioactive 137Cs decontamination
The adsorption ability of the Fe3O4@SiO2@KTiFC magnetic composite in radioactive 137Cs-spiked solutions with different initial activity was examined. The performance parameters including the decontamination factor (DF) and removal efficiency (R%) were obtained, as summarized in Table 4. It can be seen that the 137Cs activity in the solutions shows a significant decrease after contact with the particles for 90 min. The removal efficiency, which exceeded 97% under all the experimental conditions, indicates the high adsorption competence of Fe3O4@SiO2@KTiFC magnetic composite for 137Cs decontamination. It is notable that the residual activity of the 137Cs solution after treatment with Fe3O4@SiO2@KTiFC was 38 Bq L−1 at the initial concentration of 2993 Bq L−1, which was slightly higher than the background activity. In addition, high DF (>40) further confirms the application potential of the Fe3O4@SiO2@KTiFC magnetic composite for the decontamination of 137Cs-containing radioactive liquid wastes.
Table 4 Adsorption performance of Fe3O4@SiO2@KTiFC in 137Cs-spiked solutionsa
| Activity of 137Cs |
Performance parameters |
| A0 (Bq L−1) |
Af (Bq L−1) |
DF A0/Af |
R (%) |
| A0: Initial activity; Af: final activity after treatment. |
| 2993 |
38 |
79 |
98.7 |
| 5347 |
48 |
112 |
99.1 |
| 16901 |
389 |
43 |
97.7 |
| 34244 |
437 |
78 |
98.7 |
Cesium removal from seawater
As mentioned above, radionuclide-contaminated seawater in the Fukushima accident is a serious environmental concern. Because of the extremely low amount as well as the co-existence of a large number of competing metal ions, the removal of cesium in seawater remains a challenging task. In our study, real seawater from different districts of the Bohai Sea in China was employed to prepare cesium contaminated water for batch adsorption. The removal efficiency (R%) and distribution coefficient (Kd) is listed in Table 5. For the shallow-ocean seawaters with the addition of 15- or 150-fold cesium, the Fe3O4@SiO2@KTiFC magnetic composite showed a removal efficiency higher than 98%, and the Kd was higher than 104 mL g−1. After the treatment, the amount of cesium in the contaminated seawater was reduced to less than the background level. Similar results were obtained for the deep-ocean seawater, even though it had a lower concentration of cesium.
Table 5 Adsorption performance of Fe3O4@SiO2@KTiFC in cesium contaminated seawater
| Solution |
Cs(I) concentration (ppb) |
R (%) |
Kd (mL g−1) |
| Initial |
Treatment |
| Shallow-ocean water |
10.01 |
2.61 |
73.9 |
2100.2 |
| Shallow-ocean water + 0.15 ppm Cs(I) |
161.35 |
2.52 |
98.4 |
48580.8 |
| Shallow-ocean water + 1.5 ppm Cs(I) |
1603.5 |
8.28 |
99.5 |
151197.2 |
| Deep-ocean water |
2.58 |
1.23 |
52.5 |
834.8 |
| Deep-ocean water + 0.15 ppm Cs(I) |
162 |
5.46 |
96.6 |
22507.9 |
| Deep-ocean water + 1.5 ppm Cs(I) |
1660.5 |
11.25 |
99.3 |
112769.2 |
Stability evaluation in HNO3 medium
As is known, a large amount of acid co-exists with radiocesium in radioactive liquid waste. For practical application, it is essential to evaluate the acid-resistance performance of Fe3O4@SiO2@KTiFC magnetic composites in harsh acidic conditions. For this purpose, 30 mg of the magnetic composite was soaked into 20 mL of 1.0 mol L−1 nitric acid solution. After shaking for different interval times, aliquots of liquid solution were removed for the determination of the Fe3+ concentration in the aqueous phase through spectrophotometry. Fig. 8 gives the variation of the leached Fe3+ in the solution during 168 hours treatment. It can be seen that the naked Fe3O4 particles without a silica protective layer showed a fast degradation under the experimental condition. On the contrary, the Fe3O4@SiO2@KTiFC magnetic composite with a silica protective layer was stable enough under strong acid conditions. No detectable Fe3+ ions were found in the leaching solution. Therefore, it is believed that the Fe3O4@SiO2@KTiFC magnetic microspheres should be sufficiently stable for practical applications.
 |
| | Fig. 8 Acid-resistance test of the magnetite microspheres and Fe3O4@SiO2@KTiFC particles. | |
Conclusions
A novel kind of potassium titanium ferrocyanide (KTiFC) functionalized magnetic microsphere was synthesized through a core–shell architecture strategy for the removal of radiocesium. The structure, morphology, and adsorption behavior of the Fe3O4@SiO2@KTiFC magnetic composite were comprehensively investigated. The high ion exchange efficiency of KTiFC in combination of the ease of the magnetic separation provided a facile way for the cesium decontamination. The magnetic composite showed high removal efficiency even in seawater containing extremely low concentrations of cesium. Moreover, for practical application, the material has been proven to have excellent stability in strong HNO3 solutions due to the existence of silica coating as a protection layer. This magnetic composite has promising applications for the cleanup of radioactive cesium in contaminated environments.
Acknowledgements
The study was supported by Program for Changjiang Scholars and Innovative Research Team in University (IRT13026), and National Natural Science Foundation of China under Project 51103079 and 9122611.
Notes and references
- C. Liu, Y. Huang, N. Naismith, J. Economy and J. Talbott, Environ. Sci. Technol., 2003, 37, 4261 CrossRef CAS.
- H. Kawamura, T. Kobayashi, A. Furuno, T. In, Y. Ishikawa, T. Nakayama, S. Shima and T. Awaji, J. Nucl. Sci. Technol., 2011, 48, 1349 CrossRef CAS.
- R. R. Sheha, J. Colloid Interface Sci., 2012, 388, 21 CrossRef CAS PubMed.
- P. K. Mohapatra, D. S. Lakshmi, A. Bhattacharyya and V. K. Manchanda, J. Hazard. Mater., 2009, 169, 472 CrossRef CAS PubMed.
- A. Y. Zhang and Z. F. Chai, Ind. Eng. Chem. Res., 2012, 51, 6196 CrossRef CAS.
- Y. X. Leng, G. Ye, J. Xu, J. Wei, J. C. Wang and J. Chen, J. Sol–Gel Sci. Technol., 2013, 66, 413 CrossRef CAS PubMed.
- T. Sangvanich, V. Sukwarotwat, R. J. Wiacek, R. M. Grudzien, G. E. Fryxell, R. S. Addleman, C. Timchalk and W. Yantasee, J. Hazard. Mater., 2010, 182, 225 CrossRef CAS PubMed.
- P. A. Haas, Sep. Sci. Technol., 1993, 28, 2479 CrossRef CAS.
- C. Delchet, A. Tokarev, X. Dumail, G. Toquer, Y. Barre, Y. Guari, C. Guerin, J. Larionova and A. Grandjean, RSC Adv., 2012, 2, 5707 RSC.
- A. Nilchi, A. Khanchi, H. Atashi, A. Bagheri and L. Nematollahi, J. Hazard. Mater., 2006, 137, 1271 CrossRef CAS PubMed.
- V. Avramenko, S. Bratskaya, V. Zheleznov, I. Sheveleva, O. Voitenko and V. Sergienko, J. Hazard. Mater., 2011, 186, 1343 CrossRef CAS PubMed.
- H. H. Someda, A. A. ElZahhar, M. K. Shehata and H. A. El-Naggar, Sep. Purif. Technol., 2002, 29, 53 CrossRef CAS.
- C. Dwivedi, S. K. Pathak, M. Kumar, S. C. Tripathi and P. N. Bajaj, RSC Adv., 2013, 3, 22102 RSC.
- T. P. Valsala, S. C. Roy, J. G. Shah., J. Gabriel, K. Raj and V. Venugopal, J. Hazard. Mater., 2009, 166, 1148 CrossRef CAS PubMed.
- A. V. Voronina, V. S. Semenishchev, E. V. Nogovitsyna and N. D. Betenekov, J. Radioanal. Nucl. Chem., 2013, 298, 67 CrossRef CAS.
- T. Sangvanich, V. Sukwarotwat, R. J. Wiacek, R. M. Grudzien, G. E. Fryxell, R. S. Addleman, C. Timchalk and W. Yantasee, J. Hazard. Mater., 2010, 182, 225 CrossRef CAS PubMed.
- A. Lu, E. L. Salabas and F. Schüth, Angew. Chem., Int. Ed., 2007, 46, 1222 CrossRef CAS PubMed.
- H. Chen, C. Deng and X. Zhang, Angew. Chem., Int. Ed., 2010, 49, 607 CrossRef CAS PubMed.
- S. Wu, N. Duan, X. Ma, Y. Xia, Y. Yu, Z. Wang and H. Wang, Chem. Commun., 2012, 48, 4866 RSC.
- Y. Zhao, J. Li, S. Zhang, H. Chen and D. Shao, RSC Adv., 2013, 3, 18952 RSC.
- M. Ye, Q. Zhang, Y. Hu, J. Ge, Z. Lu, L. He, Z. Chen and Y. Yin, Chem.–Eur. J., 2010, 16, 6243 CrossRef CAS PubMed.
- J. Huang, R. Zhao, H. Wang, W. Zhao and L. Ding, Biotechnol. Lett., 2010, 32, 817 CrossRef CAS PubMed.
- S. Xuan, F. Wang, X. Gong, S. Kong, J. C. Yu and K. C. Leung, Chem. Commun., 2011, 47, 2514 RSC.
- H. Tan, J. M. Xue, B. Shuter, X. Li and J. Wang, Adv. Funct. Mater., 2010, 20, 722 CrossRef CAS PubMed.
- R. D. Ambashta, P. K. Wattal, S. Singh and D. Bahadur, J. Magn. Magn. Mater., 2003, 267, 335 CrossRef CAS.
- C. Thammawong, P. Opaprakasit, P. Tangboriboonrat and P. Sreearunothai, J. Nanopart. Res., 2013, 15, 1689 CrossRef.
- H. Yang, L. Sun, J. Zhai, H. Li, Y. Zhao and H. Yu, J. Mater. Chem. A, 2014, 2, 326 CAS.
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. V. Elst and R. N. Muller, Chem. Rev., 2008, 108, 2064 CrossRef CAS PubMed.
- M. D. Kaminski and L. Nuñez, Sep. Sci. Technol., 2002, 37, 3703 CrossRef CAS PubMed.
- M. F. Shao, F. Y. Ning, J. W. Zhao, M. Wei, D. G. Evans and X. Duan, J. Am. Chem. Soc., 2012, 134, 1071 CrossRef CAS PubMed.
- Y. Deng, D. Qi, C. Deng, X. Zhang and D. Zhao, J. Am. Chem. Soc., 2007, 130, 28 CrossRef PubMed.
- R. Yi, G. Ye, D. Pan, F. Wu, M. Wen and J. Chen, J. Mater. Chem. A, 2014, 2, 6840 CAS.
- M. Kaur, A. Johnson, G. X. Tian, W. L. Jiang, L. F. Rao, A. Paszczynski and Y. Qiang, Nano Energy, 2013, 2, 124 CrossRef CAS PubMed.
- X. Feng, Q. He, Z. Chen, X. Han and J. Guo, Radiat. Meas., 2011, 46, 533 CrossRef CAS PubMed.
- Q. Zhang, D. Q. Lima, I. Lee, F. Zaera, M. F. Chi and Y. D. Yin, Angew. Chem., Int. Ed., 2011, 123, 7226 CrossRef PubMed.
- X. Feng, S. Jing, Q. Wu, J. Chen and C. Song, Chin. J. Chem. Eng., 2007, 15, 184 CrossRef CAS.
- Y. S. Ho and G. McKay, Process Saf. Environ. Prot., 1998, 76, 332 CrossRef CAS.
- Y. S. Ho and G. McKay, Process Biochem., 1999, 34, 451 CrossRef CAS.
- Y. S. Ho, J. Hazard. Mater., 2006, 136, 681 CrossRef CAS PubMed.
- Y. S. Ho, Water Res., 2006, 40, 119 CrossRef CAS PubMed.
- C. Dwivedi, A. Kumar, K. K. Singh, A. K. Juby, M. Kumar, P. K. Wattal and P. N. Bajaj, J. Appl. Polym. Sci., 2013, 129, 152 CrossRef CAS PubMed.
- E. Njikam and S. Schiewer, J. Hazard. Mater., 2012, 213–214, 242 CrossRef CAS PubMed.
- F. F. Bai, G. Ye, G. J. Chen, J. C. Wei, J. C. Wang and J. Chen, React. Funct. Polym., 2013, 73, 228 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.