
Simultaneous deposition of cerium oxide and gold nanostructures-characterization and analytical properties toward glucose electro-oxidation and sensing†
Maxime Gougisa,
Antonio Pereirab,
Dongling Maa and
Mohamed Mohamedi*a
aINRS-Énergie, Matériaux et Télécommunications, 1650 Boulevard Lionel Boulet, Varennes, Québec, Canada J3X 1S2. E-mail: mohamedi@emt.inrs.ca; Fax: +1-450-929-8102; Tel: +1-514-228-6831
bInstitut Lumiére Matiére - UMR 5306, Université, Claude Bernard Lyon 1, 10 rue Ada Byron, 69622, Villeurbanne cedex, France
First published on 8th August 2014
Abstract
Here, we present for the first time an Au–CeO2 nanocomposite synthesized in the presence of a mild pressure of oxygen (1.33 Pa) by the cross-beam laser deposition technique. The deposit was grown on a conductive (current collector) substrate composed of a porous network of carbon microfibers. The structure and morphology of the deposit were characterized by X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), scanning electron microscopy (SEM), and transmission electron microscopy (TEM). XPS analysis demonstrated that the interaction of Au with CeO2 during deposition induced charge redistribution in the ceria nanoparticles (Ce3+,4+), resulting in positively charged Au species (Au3+). TEM analysis revealed that the basal planes of the carbon fibers are coated with a monolayer of interconnected nanoparticles, whereas their outer walls are covered with an ultra-thin film (∼40 nm thickness) formed of nanoparticles gathering into columns. The direct electrocatalytic oxidation of glucose in a neutral medium, such as phosphate buffered saline (pH = 7.2) solution, has been investigated with cyclic voltammetry, chronoamperometry and square-wave voltammetry. Sensitivity as high as 57.5 μA cm−2 mM−1 up to 10 mM of glucose and a low detection limit of 10 μM were obtained with square-wave voltammetry. Such electrocatalytic activity is because of the presence of cationic gold (electronic effect) and the size reduction of gold particles (size effect). This interesting analytical performance in addition to the low loading of gold (100 μg cm−2) makes the laser fabricated Au–CeO2 nanocomposite electrode potentially promising for glucose fuel cells, assessment of glucose in biological fluids, as well as in the analysis of food and beverages.
1. Introduction
The glucose electro-oxidation reaction (GER) is very important in implantable fuel cells,1 and in heterogeneous catalysis.2 The determination of glucose concentration is vital in clinical applications such as biosensors for diabetes requiring regular measurements of blood glucose levels,3–5 as well as in food processing and fermentation.6Gold is an attractive catalyst for the GER because of its excellent biological compatibility and oxidation potential in neutral and alkaline media that is more negative compared to the other metals,7 and thus has been extensively examined.4,8–10
Numerous metal oxides are inexpensive, largely available, and environmentally benign, and thus are developing as a distinctive class of electrode materials for a broad range of applications such as photoelectrochemical cells and energy storage and conversion devices.11–13 Among metal oxides, ceria is one of the cheapest and most abundant rare earth oxides. Hence, nanostructured CeO2 is one of the extensively studied in materials applications due to improvements in redox properties, oxygen transport properties and surface to volume ratio in comparison to the bulk materials.14
Combinatorial utilization of Au and CeO2 materials as a composite catalyst toward glucose GER is rather new. To the best of our knowledge, we were the first to consider this system for this electrochemical reaction. In our recent publication, we reported a single-beam pulsed laser deposition (PLD) of a layer onto layered (lol) CeO2–Au nanostructures.15 Various morphologies of CeO2 were synthesized under three different background atmospheres, i.e., vacuum, inert (He gas) and oxygen gas. The effect of the obtained morphologies (regular segments composed of clusters of particles, fluffy-like texture, lamellae-like texture) of the CeO2 layer on the electroactivity of gold (itself deposited by PLD) towards GER was presented. Among the fabricated electrodes, CeO2 synthesized in the presence of 1.33 Pa of oxygen exhibited the best electrocatalytic supporting properties to Au towards GER with a sensitivity of 44 μA cm−2 mM−1, and a detection limit of 10 μM within 0.01 and 10 mM of glucose concentration range.
In the present study, we present the synthesis and characterization of simultaneously deposited CeO2 and Au materials by cross-beam pulsed laser deposition (CBPLD) method in the presence of 1.33 Pa pressure of oxygen. The objective here is twofold: from a fundamental viewpoint it is to investigate the microstructural interactions of the co-deposited CeO2 and Au as compared with the previously developed lol architecture. From a practical viewpoint it is to seek further enhancement in the electrocatalytic activity and sensing of glucose for gold.
XRD, XPS, SEM and TEM were employed to investigate the structural properties of the Au–CeO2 composite. Cyclic voltammetry (CV), chronoamperometry and square-wave voltammetry (SWV) electrochemical were employed to study the activity towards GER conducted in phosphate buffered saline (PBS, pH = 7.2) solution containing glucose concentrations varying from 0.01 mM to 100 mM.
2. Experimental
2.1. Synthesis
Gold and CeO2 were deposited onto carbon paper (CP, Toray) by CBPLD at room temperature using a pulsed KrF excimer laser (λ = 248 nm, pulse width = 17 ns, and repetition rate = 50 Hz). A pure polycrystalline Au target (99.99%, Kurt J. Lesker Co.) and a pure polycrystalline CeO2 target (99.99%, Kurt J. Lesker Co.) were used as raw materials for the co-deposition. Prior to laser ablation, the PLD chamber was evacuated with a turbo pump (∼0.0026 Pa). The fluence was fixed to 4 J cm−2 and the target-to-substrate distance was set to 50 mm. The composite Au–CeO2 was deposited under 1.33 Pa of oxygen with 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 laser pulses. A gold loading of about 100 μg cm−2 was estimated by weighing the substrate before and after the deposition of gold with 20000 laser pulses using a single-beam PLD. During deposition, the Au and CeO2 targets were continuously rotated and translated to obtain a uniform ablation over the entire surface of the targets. A detailed description of the CBPLD setup has been provided elsewhere.16
000 laser pulses. A gold loading of about 100 μg cm−2 was estimated by weighing the substrate before and after the deposition of gold with 20000 laser pulses using a single-beam PLD. During deposition, the Au and CeO2 targets were continuously rotated and translated to obtain a uniform ablation over the entire surface of the targets. A detailed description of the CBPLD setup has been provided elsewhere.16
2.2. Material characterization
The morphologies of the Au–CeO2 structures were observed with SEM (JEOL JSM-6300F operating at 20 kV) and TEM (JEOL JEM-2100F operating at 200 kV). The samples were subjected as-produced to SEM and TEM analyses. XPS measurements were carried out via a VG Escalab 220i-XL equipped with an Al Kα source (1486.6 eV). The anode was operated at 10 kV and 20 mA and the pass energy of the analyzer was fixed at 20 eV. The samples were analyzed with a spot size of 250 × 1000 μm located approximately in the center of the sample. A survey spectrum ranging from 0 to 1300 eV was first acquired, and then higher resolution multiplex scan spectra (Au 4f, Ce 3d, O 1s, and C 1s core levels) were obtained. Quantification of the elements was performed with CasaXPS software version 2.3.15 (Casa Software Ltd.) by fitting the core level spectra to several Pseudo-Voigt functions (sum of Gaussian–Lorentzian function) after a Shirley background removal. The C 1s core level peak at 284.6 eV, resulting from hydrocarbon contaminants at the surface, was used as an internal reference. All the spectra were recalibrated with respect to the C 1s core level peak of adventitious carbon contamination.The crystalline structure of the samples was determined by X-ray diffraction (XRD) using a Bruker D8 Advance diffractometer equipped with a Cu Kα source. The diffractometer was operated at 40 kV and 40 mA. Diffractograms were carried out in the Grazing Incidence Diffraction (GID) scan mode with an incident angle of 2° and a 2θ angular step size of 0.05° with an acquisition time of 5 s per step.
2.3. Electrochemical characterization
Prior to the electrochemical measurements, the surface of the working electrode was cleaned electrochemically by potential cycling in 0.5 M H2SO4 (96% purity from Acros Organics). The electrochemical properties of the Au–CeO2 electrodes were investigated using CV, chronoamperometry and SWV in a de-aerated PBS solution (pH = 7.2, Sigma-Aldrich) with glucose (D-(+)-glucose, ACS reagent grade, Sigma-Aldrich). All the electrochemical measurements were carried out at room temperature using a three-compartment electrochemical cell using an Ag/AgCl, 3 M KCl reference electrode and a platinum coil as a counter electrode. Measurements and data acquisition were conducted with an Autolab potentiostat/galvanostat obtained from Eco Chemie.3. Results and discussion
3.1. Structural characterization
Fig. 1a is an SEM image at nominal magnification 200× showing the general features of Au–CeO2 deposited in the presence of 1.33 Pa of oxygen on carbon paper substrate. Images 1b and 1c are at nominal magnifications from 5000 to 20000× of a selected area from the entire sample. The images show an Au–CeO2 layer of rough morphology fully covering the carbon paper substrate.
 | ||
| Fig. 1 (a)–(c) SEM images at increasing magnifications of Au–CeO2 deposited onto carbon paper by CBPLD under 1.33 Pa of oxygen partial pressure. (d) Corresponding XRD pattern. | ||
The corresponding XRD profile of the Au–CeO2 composite is shown in Fig. 1d. The diagram shows peaks corresponding to the (111), (200), (220), and (311) crystallographic planes of the face centered cubic (fcc) gold (JCPDF 89-3697). In addition, the XRD profile shows the presence of diffraction peaks (111), (200), and (220) corresponding to the fcc fluorite structure of CeO2 (JCPDS file 13-1002). By using the Debye-Scherrer equation and the most intense (111) diffraction peak for Au and CeO2, the average crystallite size was estimated to be 36.9 nm and 6.2 nm for CeO2 and Au, respectively. The lattice constant of CeO2 is equal to 5.416 Å (close to theoretical calculations (5.411 Å)), whereas that of gold is 4.077 Å (close to theoretical calculations (4.080 Å)).
Fig. 2a shows the TEM image taken on one of the microfibers composing the carbon paper substrate. The high resolution TEM images show that the morphology of the Au–CeO2 deposit is a mixture of an ultra-thin layer composed of strongly interconnected nanoparticles along the basal plane of the carbon fiber (Fig. 2b) and a thicker film of about 40 nm thickness on the outer walls of the carbon fiber (Fig. 2c). A closer look shows that the thicker film assembles into the columns of nanoparticles. The HR-TEM image (Fig. 2d) shows that it is difficult to distinguish between Au and CeO2 particles. The particle average diameter estimated from Fig. 2d was 5.5 ±0.8 nm.
 | ||
| Fig. 2 TEM and HR-TEM analyses of Au–CeO2 deposited onto carbon paper by CBPLD under 1.33 Pa of oxygen partial pressure. | ||
Afterward, the XPS technique was employed to examine the surface constituent phases and to obtain further information on the interaction between CeO2 and Au. The XPS survey (not shown) presented peaks only for C, O, Ce and Au with a surface composition of 31.04, 38.08, 12.16 and 18.71 at%, respectively. Fig. 3a shows the high-resolution XPS Au 4f core level spectrum, which reveals that Au is present in more than one oxidation state. The Au 4f spectrum is composed of two doublets at energies 83.9/87.7 eV and 86/89.6 eV. The first doublet corresponds to metallic Au0 while the second oxidation state can be linked with the oxidized Au3+ species. Fitting the spectrum with two doublets gave an Au3+/Au0 ratio of 0.26. Au–CeO2 composites exhibiting ionized gold have been observed by some researchers,17–19 and this is the first time it has been observed for Au–CeO2 films prepared by CBPLD technique.
 | ||
| Fig. 3 XPS analysis of Au–CeO2 deposit. (a) High-resolution XPS spectrum in the Au 4f region and (b) High-resolution XPS spectrum in the Ce 3d region. | ||
The Ce 3d core level XPS spectrum of the Au–CeO2 composite reported in Fig. 3b consists of three 3d5/2–3d3/2 doublets characteristic of Ce4+ (CeO2) and two doublets of Ce3+.20 The multiplets are because of the spin–orbit coupling energies due to different 4f electron configuration in the final state. The results of Fig. 3 show that cerium oxide was partially reduced. The Ce3+/Ce4+ ratio obtained by calculating the sums of the areas of the Ce 3d doublets was 0.07.
3.2. Electrochemical and glucose determination studies
The synthesized Au–CeO2 nanocomposite was first characterised by CV to confirm the typical electrochemical response for Au. Fig. 4a shows the CV profile for Au–CeO2 in a de-aerated solution of 0.5 M H2SO4 at a scan rate of 50 mV s−1. The voltammetric profile shows the characteristic redox processes due to the formation of Au oxide within 1.1 V to 1.47 V potential range and a reduction peak centered at 0.94 V vs. Ag/AgCl, which confirm the cleanliness of Au in the synthesized Au–CeO2 nanocomposite. The electroactive surface area (ESA), estimated by integrating the charge consumed for reducing the Au oxides formed in the positive scan by assuming that the reduction of a monolayer of Au oxides requires 400 μC cm−2,21 was found to be 6.03 cm2, which is higher than the ESA obtained with the lol electrode structure CeO2–Au (4.30 cm2).15 | ||
| Fig. 4 CVs recorded at the Au–CeO2 electrode in an argon-saturated solution of: (a) 0.5 M H2SO4 recorded with 50 mV s−1 potential scan rate, and (b) 0.01 M PBS (pH = 7.2) containing 100 mM glucose recorded with 2 mV s−1. Arrows indicate the scan direction. | ||
The glucose electro-oxidation was first investigated at the Au–CeO2 nanocomposite electrode in a pH 7.2 PBS solution containing 100 mM glucose. Fig. 4b shows the CV obtained at a slow scan rate of 2 mV s−1 (quasi-steady state). The electrode displayed the typical response of glucose oxidation at the Au electrode.22 The peak current in the forward scan is 674 μA, which is 1.45 times larger than that delivered by the lol CeO2–Au electrode (450.45 μA).15
Afterward, slow scan CV with 2 mV s−1 was conducted in argon-saturated PBS solution containing various concentrations of glucose (0.01 mM to 100 mM) at the Au–CeO2 nanocomposite electrode (Fig. 5a). The maximum current (peak current) extracted from Fig. 5a was then plotted against glucose concentration, and the corresponding curve is shown in Fig. 5b. It can be seen that the peak current increases as the concentration of glucose increases. The current response varied linearly with glucose concentration from 0.01 mM to 10 mM with a regression equation of y = 26.7x + 4.21 (x: concentration/mM, y: current/μA cm−2) and a correlation coefficient of R2 = 0.9982. The sensitivity and the low detection limit were found to be 26.7 μA cm−2 mM−1 and 10 μM, respectively. For glucose concentrations higher than 10 mM, a non-linear dependence was observed because of the saturation of active adsorption sites with glucose.
 | ||
| Fig. 5 (a) Voltammetry measurements recorded with 2 mV s−1 at the Au–CeO2 electrode in argon-saturated 0.01 M PBS solution at pH 7.2 containing glucose concentrations varying from 0.01 mM to 100 mM. (b) Dependence of the peak current over the entire range of glucose concentration. Arrows indicate the scan direction. | ||
Chronoamperometry was also employed as a detection method. Fig. 6a shows the chronoamperometric measurements of glucose using the Au–CeO2 nanocomposite electrode. This figure represents the current-time profiles obtained by setting the working electrode at +0.25 V vs. Ag/AgCl for various concentrations of glucose. The stationary current signals recorded after 60 s depended linearly on glucose concentration up to 10 mM of glucose (Fig. 6b) with a regression equation of y = 25.2x + 2.86 (x: concentration/mM, y: current/μA cm−2) and a correlation coefficient of R2 = 0.9973. The sensitivity and the low detection limit were found to be 25.2 μA cm−2 mM−1 and 10 μM, respectively. These results are very similar to those obtained with voltammetry.
 | ||
| Fig. 6 (a) Chronoamperograms recorded at an applied potential of 0.25 V vs. Ag/AgCl at the Au–CeO2 electrode in argon-saturated 0.01 M PBS solution at pH 7.2 containing glucose concentration varying from 0 mM to 100 mM. (b) Dependence of the steady-state current density over the entire range of glucose concentration. | ||
Square-wave voltammetric techniques are widely used to improve the electroanalytical parameters for the direct measurement of concentration, e.g., the lowest limit of detection and sensitivities. Fig. 7a shows SWV responses recorded with the Au–CeO2 nanocomposite electrode for various concentrations of glucose under optimized conditions, i.e., frequency of 20 Hz, square-wave amplitude of 20 mV, and step potential of 1 mV. Fig. 7b shows the analytical curve, i.e., the dependence of peak current density on glucose concentration for the interval between 0.01 and 100 mM glucose. The current response varied linearly with glucose concentration from 0.01 mM to 10 mM with a regression equation of y = 57.5x + 8.86 (x: concentration/mM, y: current/μA cm−2) and a correlation coefficient of R2 = 0.9971. The sensitivity was found to be 57.5 μA cm−2 mM−1 and a low detection limit 10 μM was obtained. The sensitivity obtained with the SWV technique is twice that of those obtained by CV and chronoamperometry, and the acquisition time is also considerably shorter with the former technique. The linearity from 0 to 10 mM is far beyond the physiological level (3–8 mM). Further work will be performed to investigate the interference species, such as uric acid and ascorbic acid, in real samples envisaging practical glucose analysis.
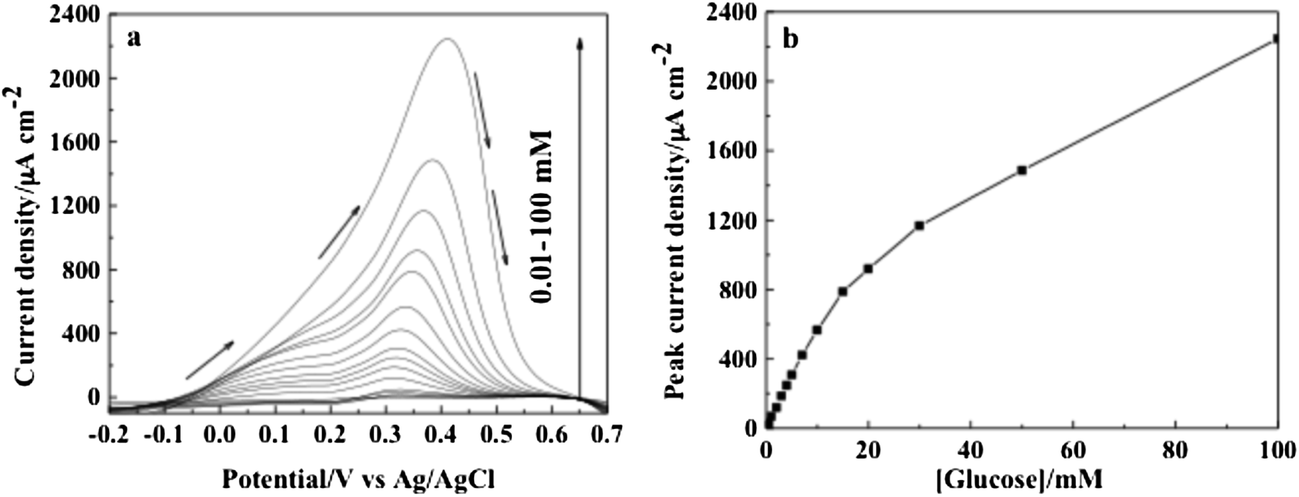 | ||
| Fig. 7 (a) SWV recorded with an amplitude of 20 mV, a step potential of 1 mV and a frequency of 20 Hz at the Au–CeO2 electrode in argon-saturated 0.01 M PBS solution at pH 7.2 containing glucose concentration varying from 10 μM to 100 mM. (b) Dependence of the peak current over the entire range of glucose concentration. Arrows indicate the scan direction. | ||
3.3. Comparison with other laser synthesized Au based electrodes
The PLD technique is one of the most suitable techniques for the synthesis of model materials for fundamental studies because the technique allows for robust control of the morphology and the dimensions as well as the retaining of stoichiometry of the target in the deposited film.23 In addition, ultra-low loading together with planar deposition are two other essential characteristics that could make PLD promising as a future-manufacturing route for on-chip miniaturized biomedical devices including analytical sensors.It is also well-known that the method of synthesis enormously affects the performance of a material. Therefore, it would be more appropriate to compare the performance of gold-based electrodes produced by the same method of synthesis at least from a fundamental standpoint. Hence, Table 1 compares sensitivity performances obtained by the SWV of gold toward glucose with different catalytic supports (carbon paper (CP), carbon nanotubes (CNTs) and CeO2). In all the cases, gold and CeO2 were synthesized by the PLD technique. In light of the results shown in Table 1, it is clear that CeO2 brings an improved effect to gold vis-à-vis the GER as compared to CP or CNTs supports. Table 1 further shows that within the two architectures of the CeO2 and Au system, the lol architecture delivered an electroactivity of gold toward oxidative sensing of glucose lower than the simultaneously deposited Au–CeO2 nanocomposite (this work). This shows the importance of the designing of the Au–CeO2 architecture to fully benefit from the promoting properties of CeO2.
XPS analysis of the lol CeO2–Au structure did not show a change in the electronic structure of Au deposited onto the CeO2 layer in the presence of 1.33 Pa of oxygen.15 On the contrary, XPS shown in this work revealed that when CeO2 is simultaneously deposited with Au, the latter is partially oxidized to Au3+ and Ce4+ is moderately reduced to Ce3+. It should be noted that we conducted the co-deposition of Au and CeO2 by CBPLD under vacuum and under an inert atmosphere of helium gas; no cationic gold was detected by XPS (Fig. S1†). Thus, it can be expected that PLD deposition in an atmosphere of oxygen would result in the dissociation of O2 molecules and the production of ionized oxygen, which would react with gold to form an oxide.26 However, such mechanism was observed only for oxygen pressures higher than 13.33 Pa.25,27 Similarly, in the case of CeO2 and Au co-deposited under 1.33 Pa of oxygen partial pressure, a part of this oxygen dissociates and ionizes due to the laser during deposition and there is added ionized oxygen obtained from the CeO2 target. Consequently, the proportion of oxygen is larger during the deposition, which enables the occurrence of gold oxide. Therefore, despite the concentration of Au3+ being low, its presence in the co-deposited Au–CeO2 sample appears to have contributed to the enhancement of electroactivity towards GER as compared to the CeO2–Au lol structure.
However, the presence of gold ions cannot be solely responsible for the good electrochemical performance of the co-deposited CeO2–Au nanocomposites. Indeed, the size of the crystallites of gold, which is another obvious parameter with a more positive impact on the activity toward GER. This can be understood by Table 1, which summarizes the crystallite sizes of gold determined by XRD with different catalyst supports and electrode structures. As can be seen, the co-deposition of CeO2 with Au yielded crystallite sizes of the latter considerably smaller than the other electrode structures. This indicates that CeO2 affects the dispersion quality of Au nanoparticles. We conducted control syntheses by co-depositing Au and CeO2 by CBPLD under vacuum or inert gas (He), and obtained an average crystallite size for Au of 8 nm and 6 nm, respectively (Fig. S2†). Therefore, small crystallite sizes of Au are only obtained when CeO2 and Au are simultaneously deposited irrespective of the background atmosphere in the deposition chamber. It is known that as the grain size reduces, the reactive sites increase and exhibit enhanced catalytic activity.28
A further improvement of the Au–CeO2 electrode is that it contains a smaller amount of Au (100 μg cm−2) as compared to the CeO2–Au lol architecture (265 μg cm−2), which is appealing for commercial applications considering the cost of gold.
4. Conclusion
The CBPLD method is employed for the first time to synthesize a Au–CeO2 nanocomposite on a substrate composed of a porous network of carbon microfibers. The synthesis was performed in the presence of a mild pressure of oxygen (1.33 Pa) in the deposition chamber. TEM analysis showed that the basal planes of the carbon fibers were coated with a monolayer of strongly interconnected nanoparticles, whereas their outer walls were coated with an ultra-thin film (∼40 nm) formed of nanoparticles assembling into columns. Ceria particles possessed Ce both in the 3+ and 4+ oxidation states as detected by XPS with very low concentrations of the former. The gold deposit exhibited a polycrystalline structure and revealed a low concentration of Au3+ ions. These results suggest that the interaction of Au with CeO2 in the presence of oxygen involves charge redistribution in the ceria nanoparticles, resulting in positively charged Au species.The analytical application of such composites towards the oxidation and detection of glucose was investigated using CV, chronoamperometry and SWV techniques. A sensitivity as high as 57.5 μA cm−2 mM−1 was obtained with SWV up to 10 mM of glucose and a low detection limit of 10 μM. This performance constitutes a very good enhancement as compared with the layer onto layer Au–CeO2 architecture and carbon support-based gold electrodes due to the presence of cationic gold (electronic effect) and to the size reduction of gold particles (size effect).
This interesting analytical performance makes the laser fabricated Au–CeO2 nanocomposite electrode potentially promising for glucose fuel cells, biomedical analysis (assessment of glucose in biological fluids, such as blood, saliva, sweat, urine, and serum), as well as for food and beverage analysis.
Acknowledgements
This work was financially supported by the Natural Sciences Engineering Research Council of Canada (NSERC), and the Fonds Québécois pour la Recherche en Nature et Technologie (FQRNT).References
- F. S. Saleh, L. Mao and T. Ohsaka, Analyst, 2012, 137, 2233–2238 RSC.
- V. Choudhary, A. B. Pinar, R. F. Lobo, D. G. Vlachos and S. I. Sandler, ChemSusChem, 2013, 6, 2369–2376 CrossRef CAS PubMed.
- B. Šljukic, C. E. Banks, C. Salter, A. Crossley and R. G. Compton, Analyst, 2006, 131, 670–677 RSC.
- K. E. Toghill and R. G. Compton, Int. J. Electrochem. Sci., 2010, 5, 1246–1301 CAS.
- C. Hou, Q. Xu, L. Yin and X. Hu, Analyst, 2012, 137, 5803–5808 RSC.
- R. Monosik, M. Stredansky, J. Tkac and E. Sturdik, Food Anal. Methods, 2012, 5, 40–53 CrossRef.
- M. Pasta, F. La Mantia and Y. Cui, Electrochim. Acta, 2010, 55, 5561–5568 CrossRef CAS PubMed.
- J. Wang, Microchim. Acta, 2012, 177, 245–270 CrossRef CAS.
- S. Park, H. Boo and T. D. Chung, Anal. Chim. Acta, 2006, 556, 46–57 CrossRef CAS PubMed.
- C. Chen, Q. Xie, D. Yang, H. Xiao, Y. Fu, Y. Tan and S. Yao, R. Soc. Chem. Adv., 2013, 3, 4473–4491 CAS.
- K. Singh, J. Nowotny and V. Thangadurai, Chem. Soc. Rev., 2013, 42, 1961–1972 RSC.
- N. Nuraje, R. Asmatulu and S. Kudaibergenov, Curr. Inorg. Chem., 2012, 2, 124–146 CrossRef CAS.
- Y. Ren, Z. Ma and P. G. Bruce, Chem. Soc. Rev., 2012, 41, 4909–4927 RSC.
- C. Sun, H. Li and L. Chen, Energy Environ. Sci., 2012, 5, 8475–8505 CAS.
- M. Gougis, A. Tabet-Aoul, D. Ma and M. Mohamedi, Microchim. Acta, 2014, 181, 1207–1214 CrossRef CAS.
- A. Tselev, A. Gorbunov and W. Pompe, Rev. Sci. Instrum., 2001, 72, 2665–2672 CrossRef CAS PubMed.
- V. Matolín, M. Cabala, I. Matolínová, M. Škoda, J. Libra, M. Václavu, K. C. Prince, T. Skála, H. Yoshikawa, Y. Yamashita, S. Ueda and K. Kobayashi, J. Phys. D: Appl. Phys., 2009, 42, 115301 CrossRef.
- S. A. C. Carabineiro, A. M. T. Silva, G. Dražić, P. B. Tavares and J. L. Figueiredo, Catal. Today, 2010, 154, 21–30 CrossRef CAS PubMed.
- M. Wang, F. Wang, J. Ma, M. Li, Z. Zhang, Y. Wang, X. Zhang and J. Xu, Chem. Commun., 2014, 50, 292–294 RSC.
- V. Matolín, M. Cabala, V. Cháb, I. Matolínová, K. C. Prince, M. Škoda, F. Šutara, T. Skála and K. Veltruská, Surf. Interface Anal., 2008, 40, 225–230 CrossRef PubMed.
- H. Angerstein-Kozlowska, B. E. Conway, A. Hamelin and L. Stoicoviciu, J. Electroanal. Chem., 1987, 228, 429–453 CrossRef CAS.
- T. M. Cheng, T. K. Huang, H. K. Lin, S. P. Tung, Y. L. Chen, C. Y. Lee and H. T. Chiu, ACS Appl. Mater. Interfaces, 2010, 2, 2773–2780 CAS.
- D. B. Chrisey and G. K. Hubler, Pulsed Laser Deposition of Thin Films, Wiley, New York, 1994 Search PubMed.
- M. Gougis, A. Tabet-Aoul, D. Ma and M. Mohamedi, Sens. Actuators, B, 2014, 193, 363–369 CrossRef CAS PubMed.
- M. Gougis, A. Pereira, D. Ma and M. Mohamedi, Int. J. Electrochem. Sci., 2014, 9, 3588–3601 CAS.
- A. Camposeo, F. Cervelli, F. Fuso, M. Allegrini and E. Arimondo, Appl. Phys. Lett., 2001, 78, 2402–2404 CrossRef CAS PubMed.
- E. Irissou, M.-C. Denis, M. Chaker and D. Guay, Thin Solid Films, 2005, 472, 49–57 CrossRef CAS PubMed.
- R. van Hardeveld and F. Hartog, Adv. Catal., 1972, 22, 75–113 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra05374e |
| This journal is © The Royal Society of Chemistry 2014 |