Reduction of CuO nanowires confined by a nano test tube
Lu Yuana,
Abram G. Van Der Geestb,
Wenhui Zhua,
Qiyue Yina,
Liang Lia,
Aleksey N. Kolmogorovb and
Guangwen Zhou*a
aDepartment of Mechanical Engineering and Multidisciplinary Program in Materials Science and Engineering, State University of New York, Binghamton, NY 13902, USA. E-mail: gzhou@binghamton.edu
bDepartment of Physics, State University of New York, Binghamton, NY 13902, USA
First published on 30th June 2014
Abstract
Using in situ transmission electron microscopy observations of the thermally induced reduction of CuO nanowires sheathed by a carbon shell, we show that a confined nanoscale geometry leads to changes in the oxide reduction mechanism from a surface dominated process to the bulk dominated process. It is shown that the reduction of carbon-confined CuO nanowires occurs via oxygen vacancy clustering in the bulk that results in the nanowire fragmentation into Cu2O segments encapsulated by the carbon shell while the reduction of un-confined CuO nanowires proceeds via the nucleation and growth of Cu2O islands on the nanowire surface. The comparative in situ TEM observations demonstrate that the surface coating layer reduces the thermal stability of the oxide nanowires, which is in contrast to the commonly anticipated effect of enhancing the nanostructure stability by developing a surface protective coating layer. Our density functional theory analyses reveal that the effects of oxygen vacancy ordering at the surface and in the bulk of CuO are comparable in energy, which support the alternative reduction process observed in the bulk of the sheathed CuO nanowires.
1. Introduction
Metal oxides are of great importance to a large variety of chemical and materials applications ranging from catalysis to electronic devices. The reduction of metal oxides, a reaction of removing lattice oxygen, plays a crucial role for these applications.1–3 For instance, pure stoichiometric oxides usually do not exhibit high catalytic activity and oxide reduction is frequently employed to modify their adsorption properties.4–9 Other processes of oxide reduction include fabrication of electronic devices, magnetic memory components, active/passive solar materials, solid-oxide fuel cells and oxygen separation membranes, where metal oxides are used as working materials.10–14 Traditionally, the reduction of metal oxides has been described using the nucleation and growth model or the interface model.2,3,15,16 In the nucleation and growth model, generation of small nuclei of the new phase (a lower oxide) occurs on the parent oxide and the reaction interface area increases until growing nuclei coalesce and then decreases.2,15–18 In the interface model, the rapid formation of a uniform and continuous layer of the reduced phase on the parent oxide occurs and the reaction boundary moves inward as the reaction proceeds.2,15–17,19 Although these phenomenological models have been found useful in the description of the reduction of bulk oxides,15,16,18,20–22 here we show that they do not apply to the reduction of confined CuO nanowires. Our in situ TEM observations of the reduction of carbon-sheathed CuO nanowires reveal that the reduction of the geometrically confined oxides follows an internal reaction process initiated by forming oxygen vacancies in the bulk rather than on the surface, which deviates significantly from the surface-dominated reduction mechanisms assumed by the aforementioned phenomenological models. The results also unravel a unique feature of the surface confinement effect on the oxide reduction – modifying the bulk stoichiometry, which is typically limited to the surface region for unconfined surfaces.CuO nanowires are chosen for our study because the nanowire morphology presents a highly anisotropic structure and it is of fundamental and technological interest to understand how oxide-reduction induced physical transformations take place within one-dimensional systems. In this context, the reduction of one-dimensional oxide nanostructures may lead to substantial changes in size, shape, and reaction intermediates provided that the nanoscale systems are free to evolve in volume and surface area. We use a volume-restricting carbon shell as a nano test tube to examine the effect of the geometrical confinement on the reaction morphology and pathway. Using in situ transmission electron microscopy (TEM), we deposit a rigid carbon coating layer around a CuO nanowire and image it as it is being reduced upon heating over 400 °C. Using this method, the reduction pathway and reaction morphology can be measured without significant change in reaction volume throughout the reduction process. By comparing with the reduction behavior of un-confined CuO nanowires, we find that the surface confinement leads to a reduction process that does not follow either the nucleation and growth mechanism or the interface mechanism.
Among many metal oxides, the reduction of copper oxides is an important reference system for understanding the reduction mechanism.14–16,20,21,23–28 Cu oxides form three distinct phases CuO (cupric), Cu2O (cuprous), and Cu4O3 (paramelaconite). In addition to being a long-debated question in solid-state chemistry, the existence of the suboxides during the reduction of CuO is closely related to the ongoing quest for the active oxide phase in heterogeneous catalysis such as the water–gas shift reaction, methanol synthesis, and methanol oxidation,29–34 where the reduction of copper oxides is frequently involved. Here we show that the thermally driven reduction of CuO nanowires results in the formation of Cu2O without involving the intermediate phase of Cu4O3. We further demonstrate that the carbon-sheathed CuO nanowires show less stability toward the thermal reduction compared to the unsheathed CuO, i.e., the sheathed CuO nanowires cannot withstand the same high temperature as the unsheathed CuO nanowires for maintaining the one-dimensional nanowire morphology, which is in contrast to the commonly expected effect of enhancing the stability of nanostructures by developing a surface coating layer.
2. Experimental and theoretical approaches
The CuO nanowires used for the reduction experiments were prepared by oxidizing a polycrystalline Cu foil (99.99% purity, obtained from Sigma-Aldrich) at 450 °C for 2 h in a vacuum chamber filled with oxygen gas with the pressure of 200 Torr. This yields well-aligned CuO nanowires perpendicular to the Cu substrates.35–37 For TEM imaging of the reduction process, CuO nanowires removed from the Cu substrate were suspended in ethanol with ultrasonication for 5 min and then drop cast onto a lacey carbon TEM grid, which was mounted onto a Gatan heating holder with rapid heating capability using a Gatan hot stage temperature controller. The TEM holder was loaded into a JEOL JEM2100F TEM. In order to confine the nanowire, carbon was deposited onto the CuO nanowires under TEM electron beam illumination during the heating process. Carbon shell formation induced by electron beam irradiation in the TEM is well-known to occur as a result of interaction between the electron beam and hydrocarbons adsorbed on the electron bombarded surface.38–40 To examine if the carbon coating layer plays any chemical effect on the oxide reduction, a Sundew atomic-layer deposition (ALD) system was also employed to deposit a thin amorphous Al2O3 layer on CuO nanowires for comparing the reduction morphology and products.Concurrent with the experimental observations, we have carried out ab initio calculations to compare the energetics of CuO reduction at surface and in the bulk. It has been widely discussed41,42 that the description of copper oxides requires careful treatment of the strongly correlated effects for partially filled Cu 3d states with DFT + U43 or hybrid functionals, such as HSE06.44 We employ DFT + U (with the Perdew–Burke–Ernzerhof functional45 and the previously selected U–J value of 6.52 eV (ref. 46)) since this efficient method has been shown to give reliable energetics of VO formation (with a small overestimation by 0.2–0.4 eV/VO compared to the HSE06 values).42 We have used a 500 eV cut-off in all calculations including ionic relaxations and full unit cell optimizations. 8 × 8 × 8 and 5 × 5 × 1 k-point meshes have been generated for the bulk and the (111) surface calculation of the CuO phase, respectively, to ensure numerical convergence of relative energies to within 2–3 meV per atom. We have used spin polarized calculations to account for the known antiferromagnetic ordering in Cu–O phases.47 The magnetic moments were found to be close to ±0.71 μB for 4-fold coordinated Cu2+ cations in the simulated CuO and derived structures. In calculations of the (111) CuO surface we have fixed the 2 × 1 base with DFT + U-optimized lattice parameters a = 5.852 Å, b = 6.219 Å, and γ = 102.74 and separated the slabs with at least 12 Å of vacuum. All surface and vacancy calculations included full ionic relaxations. The resulting (111) CuO surface energy of 0.74 J m−2 agrees well with the previously reported 0.72–0.74 J m−2 values obtained with DFT + U.42,48 The calculation of vacancy formation energies, EVO = ECumOn−1 − ECumOn + μO, requires a proper choice of the elemental chemical potentials μO. The T = 0 K energy for an isolated O2 molecule calculated with DFT is typically adjusted with a P and T-dependent term to account for the entropy of O in the gaseous state as discussed in ref. 42. Fortunately, our study focuses primarily on the evaluation of the difference in the VO formation at the surface and in the bulk which does not require μO.
3. Results and discussion
Fig. 1 shows a time-sequential series of in situ TEM images of a CuO nanowire during the heating process. As seen in Fig. 1(a), the straight CuO nanowire has a uniform diameter of ∼100 nm with smooth surface. A thin layer of amorphous carbon was developed around the oxide nanowire at room temperature due to the electron bean irradiation. The uniform TEM contrast suggests that the nanowire has a single crystalline structure without significant structural defects. With increasing the temperature, the carbon layer thickened and reached a final thickness of ∼350 nm at the bulge area at ∼260 °C. Further increase in temperature resulted in no obvious change in the thickness of the carbon coating layer. When the temperature reached 266 °C, the TEM contrast within the nanowire became non-uniform. This feature became more obvious at the higher temperature as shown in Fig. 1(e), where the visible voids are marked by red circles. By comparing Fig. 1(e and f), one can see that the void marked by the smaller red circle in Fig. 1(e) disappeared at the higher temperature, suggesting there was massive atom migration during the oxygen release. Increasing the temperature to 427 °C resulted in drastically enhanced reaction kinetics. Fig. 1(g and h) reveal that the voids started to merge, and within just one minute, the long nanowire became fragmented, resulting in a large gap between the fragmented segments.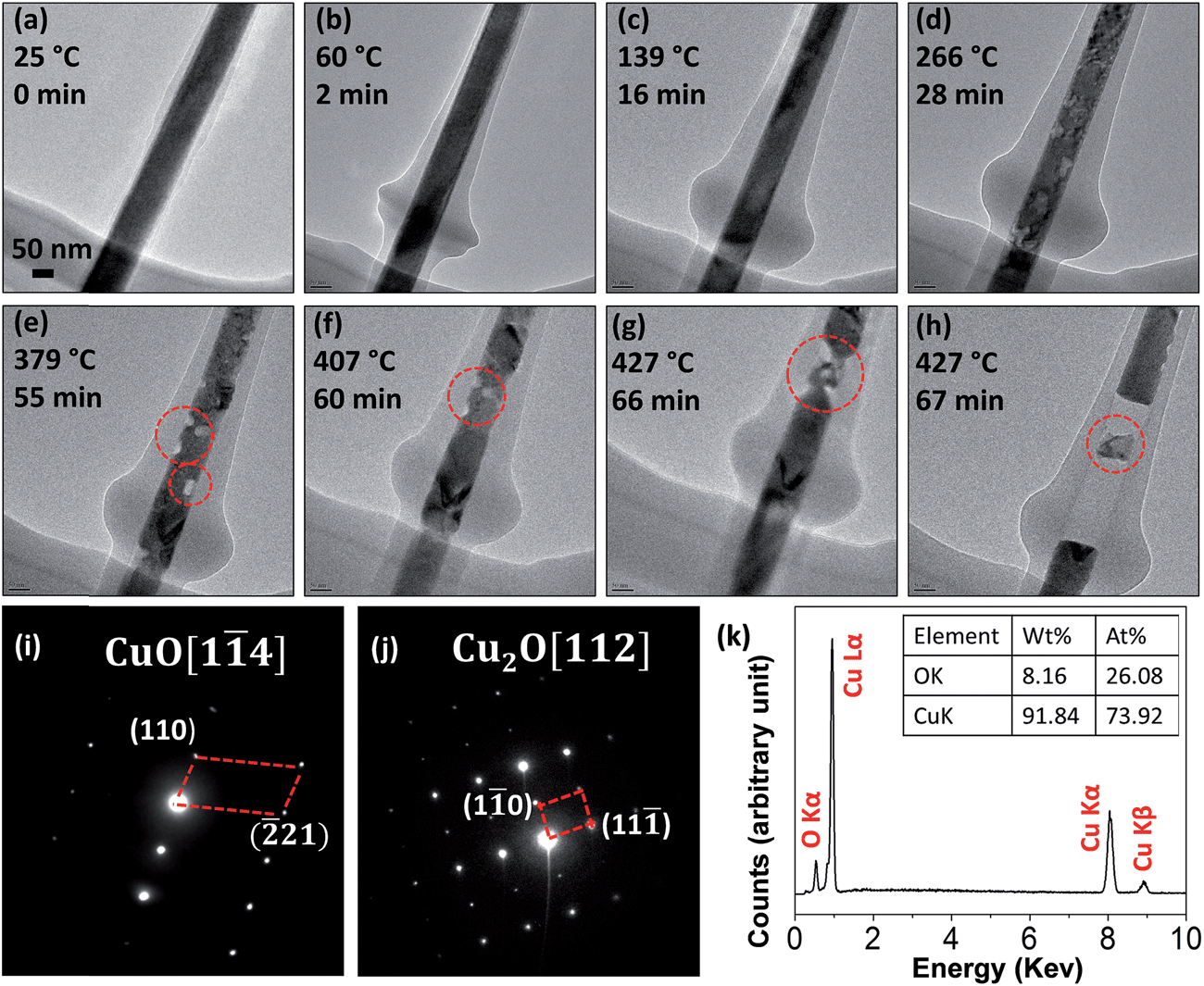 | ||
| Fig. 1 (a–h) Time sequential in situ TEM images of a single CuO nanowire as it was heated. The carbon layer increased in thickness initially and reached its final thickness at 266 °C. The oxide reduction occurs initially via losing oxygen thereby forming oxygen vacancies in the bulk while still retaining the CuO lattice structure, followed by fragmentation into Cu2O segments at ∼427 °C. (i) SAED pattern from the circle area marked in (g); (j) SAED pattern taken from the nano segment indicated by the dashed red circle in (h); (k) EDS result from the area marked by the dashed red circle in (h). | ||
Fig. 1(i) is a selected area electron diffraction (SAED) pattern from the nanowire before the fragmentation (i.e., Fig. 1(f)), which reveals that the oxide nanowire still has the CuO structure. Fig. 1(j) is a SAED pattern from the small segment formed from the fragmentation as indicated by the red circle in Fig. 1(h) and its indexing matches well with the crystal structure of Cu2O. Fig. 1(k) is an X-ray energy dispersive spectrum (EDS) from the same area indicated by the red circle in Fig. 1(h), which confirms that the segment contains both Cu and O and their atomic ratio is close to Cu2O. The in situ TEM observation reveals that the CuO nanowire is reduced to Cu2O by an abrupt fragmentation process without involving the nucleation and growth of Cu2O particles.
As described earlier, the carbon shell formation is induced by the electron beam irradiation. This can be further confirmed by a zoomed-out TEM view of a reduced sample. Fig. 2(a) shows a low-magnification TEM image showing the overall view of the sample after the thermal reduction at the peak temperature of 427 °C, where a portion (right-hand side) of the nanowire within the red dashed rectangle was originally illuminated by the TEM electron beam that resulted in the formation of a carbon shell while the rest of the sample area was outside of the TEM illumination. One can see that the nanowires without the TEM electron-beam illumination maintained their bare surface (i.e., no carbon shell formation) and showed a completely different reaction morphology compared to the electron-beam illuminated area. As indicated by dashed red circles in Fig. 2(a), small bulges were formed on the surface of bare nanowires after the thermal reduction. Fig. 2(b) is a zoomed-in TEM image from the area indicated by the red dashed rectangle in Fig. 2(a), where the lower-right hand area was continuously illuminated by TEM electron beam during the reduction while the upper-left hand corner was originally outside of the electron beam illumination. It can be seen that the nanowire illuminated by the electron beam developed a thick carbon shell and fragmented into two Cu2O segments as identified by electron diffraction analysis, consistent with the result shown in Fig. 1. The CuO nanowire outside the electron beam irradiation during the heating process was reduced to form bulges without clear fragmentation. Fig. 2(c) shows a representative electron diffraction pattern obtained from the small bulges formed on the unsheathed CuO nanowires (i.e., no carbon shell), as indicated by the red circles shown in Fig. 2(a), which reveals that they are Cu2O particles. Fig. 2(d) shows the EDS measurement of the bulged areas, which indicates that the particles contain both Cu and O. The EDS composition analysis shown in Fig. 2(d) indicates that the atomic ratio of Cu to O is 2.79![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, which is greater than the stoichiometric ratio of 2:1 for a perfect Cu2O structure. This suggests that the oxide reduction resulted in a large amount of oxygen vacancies in the Cu2O particle due to the removal of lattice oxygen.
:1, which is greater than the stoichiometric ratio of 2:1 for a perfect Cu2O structure. This suggests that the oxide reduction resulted in a large amount of oxygen vacancies in the Cu2O particle due to the removal of lattice oxygen.
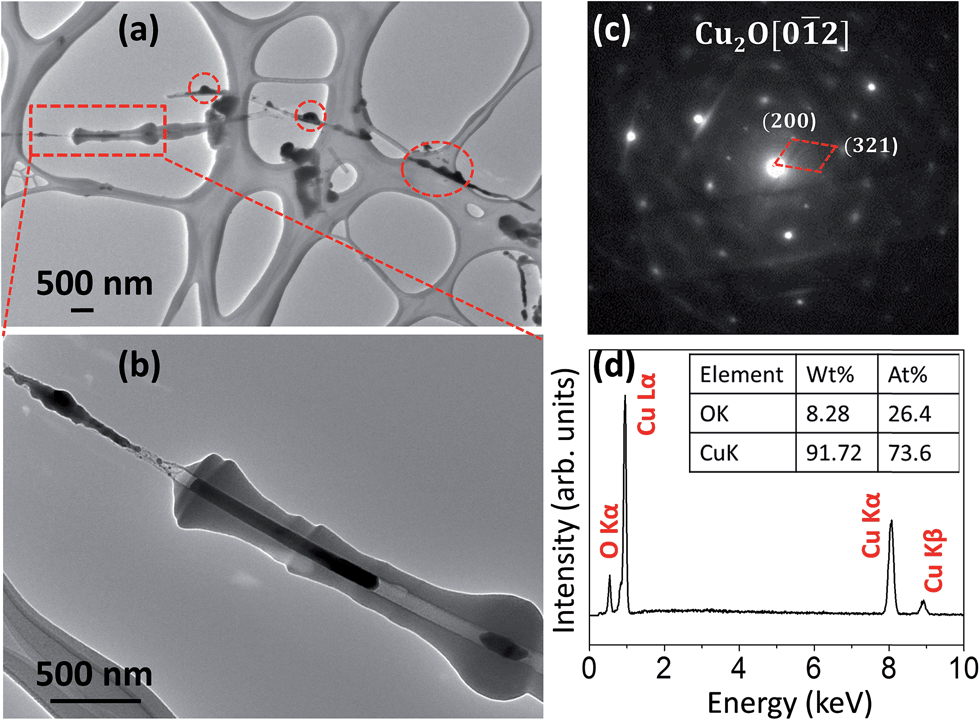 | ||
| Fig. 2 (a) A low magnification bright field TEM image showing the overview of the sample after the reduction at ∼427 °C, where a portion (left-hand side) of the CuO nanowire indicated by the red-dashed rectangular box was continuously illuminated by TEM electron-beam to form a carbon shell while the rest of the sample was outside of the TEM electron-beam illumination. (b) A closer TEM view from the red-dashed rectangular area indicated in (a); (c) a representative SAED pattern and (d) EDS from the bulges indicated by red-dashed circles in (a). | ||
The TEM observations above reveal the dramatically different reaction morphologies for carbon-sheathed CuO nanowires and unsheathed CuO nanowires. Carbon-sheathed CuO nanowires were reduced to Cu2O by fragmentation while unsheathed CuO nanowires were reduced to Cu2O by forming bulges on the nanowire surface. To further verify their difference, we used plasma cleaning (Solarus, Model 950) to remove adsorbed hydrocarbon on the as-prepared CuO nanowires in ethanol, and then performed the similar in situ TEM heating experiment. Fig. 3 shows time-sequential in situ TEM images of the morphology transformations of a plasma-cleaned CuO nanowire during the in situ heating process. One can see clearly that there was no carbon shell formation around the oxide nanowire during the entire heating process. Meanwhile, the nanowire had no significant morphology change until the temperature reached 327 °C, at which the CuO nanowire surface became roughened (Fig. 3(b)) compared to the initially smooth surface. When the temperature reached 397 °C, a small round-shaped particle became visible on the CuO nanowire surface (Fig. 3(c)). The particle grew larger while the diameter of the CuO nanowire shrank with increasing reduction temperature. At the temperature of 444 °C, the particle size grew to ∼160 nm but the nanowire diameter shrank to ∼120 nm from its original diameter of 160 nm (Fig. 3(d)). At 449 °C, another small particle appeared visible on the CuO nanowire surface as shown in Fig. 3(e). The two particles kept growing and gradually coalesced upon the continued CuO reduction (Fig. 3f–h). Fig. 3(i) is an SAED pattern obtained from the merged particle as indicated by the dashed red circle in Fig. 3(h). Indexing of the diffraction pattern matches well with the Cu2O structure. This also confirms the result shown in Fig. 2, i.e., unsheathed CuO nanowires were reduced to Cu2O by forming Cu2O bulges. Compared to the reduction of carbon-sheathed CuO nanowires that results in fragmented segments of Cu2O at the peak temperature of 427 °C with a total annealing time of 67 min (Fig. 1), reduction of the unsheathed CuO nanowire occurs via nucleation and growth of Cu2O bulges on the nanowire surface without fragmenting the original nanowire, even after being reduced at a higher peak temperature (T = 462 °C) while almost doubling the annealing time (128 min). This demonstrates that the unsheathed CuO nanowires are more stable than the carbon sheathed CuO nanowire in terms of maintaining the one-dimensional nanowire morphology.
 | ||
| Fig. 3 (a–h) Time sequential in situ TEM images taken from a plasma-cleaned CuO nanowire as it was heated; (i) SAED pattern obtained from the round area marked in (h); (j) Size evolution of the two Cu2O particles measured from the in situ TEM observations. | ||
As more O atoms leave the oxide lattice, there are more oxygen vacancies in the oxide. Therefore, it is reasonable to expect that the diffusion rates of Cu and O increase with reduction time. As a result, the oxide reduction kinetics can be autocatalytic. To check if the oxide reduction is indeed autocatalytic, we measured the size evolution of the two Cu2O particles in the course of the reduction of the CuO nanowire from the in situ TEM observation as shown in Fig. 3(a–h). Fig. 3(j) shows the measured sizes of the two Cu2O particles as a function of reduction time, which exhibits a typical sigmoid curve of an autocatalytic reaction, i.e., the rate of reaction is low in the beginning, accelerates thereafter, and then tapers off as the reactants are consumed. Note that the incubation time for the small Cu2O particle is not as significant as that for the large particle, this difference may be related to the effect from the growth of the large neighboring Cu2O particle that has already resulted in a large amount of oxygen vacancies, which may facilitate the growth of the small Cu2O particle without involving the initial slow growth stage.
Since both the in situ TEM experiments as shown in Fig. 1 and 3 were performed under the similar heating and electron-beam irradiation conditions, it is reasonable to conclude that their difference in the reduction morphologies between the sheathed and unsheathed CuO nanowires originates from the volume confinement effect exerted by the carbon sheath rather than from the electron beam irradiation effect. The presence of the carbon shell modifies the nucleation and growth behavior of the lower oxide during the reduction. As seen in Fig. 3, the reduction of unsheathed CuO nanowires results in the nucleation of Cu2O islands that grow three-dimensionally into the free space. The in situ TEM observations indicate that the CuO → Cu2O transition is localized via migration of Cu and O atoms from other regions of the CuO nanowire to the nucleated Cu2O islands, which results in the surface roughening and diameter shrinkage of the nanowire as seen in Fig. 3.
For carbon-sheathed CuO nanowires, the amorphous carbon coating layer serves as a scabbard of the nanowire and suppresses the nucleation and growth of 3D Cu2O bulges that requires free space to accommodate the local volume increase. Alternatively, due to the volume confinement by the carbon shell, the oxide reduction occurs via oxygen vacancy clustering in the bulk. Therefore, the overall CuO lattice structure of the nanowire is still maintained during the process of losing oxygen, as known from the electron diffraction analysis of the nanowire at the intermediate temperatures (Fig. 1(g and i)). With the continued loss of oxygen, the CuO nanowire is supersatured with oxygen vacancies and becomes increasingly unstable, which leads to an abrupt collapse of the CuO structure to form the more stable Cu2O structure at the peak temperature of 427 °C. Such a process is dramatically different from the reduction of unsheathed CuO nanowires, which occurs via releasing oxygen from the outer surface (as evidenced by the continued shrinkage of the nanowire diameter and formation of Cu2O islands on the nanowire surface), the un-reacted zone of the unsheathed nanowire can still maintain its intact CuO lattice structure without forming significant oxygen vacancies in the bulk. This explains why the unsheathed CuO nanowires can survive at a higher peak temperature with a much longer reduction time because the reduction occurs on the surface and the unreacted zone is not saturated with oxygen vacancies, thereby making the unsheathed structure more stable compared to the carbon-sheathed CuO nanowire.
The amorphous carbon thin film formed by electron-beam induced deposition shows a hardness of ∼4 GPa and an elastic modulus of 30–60 GPa.38 Such amorphous carbon thin films are usually used as the clamps to hold nanomaterials to certain positions.49,50 Therefore, the carbon shell on CuO nanowires works as a hard scabbard, making the formation of Cu2O bulges unfavorable. Therefore, the reduction of carbon-sheathed CuO nanowires occurs via forming oxygen vacancies in the bulk with the loss of oxygen. This leads to the collapse of the CuO structure to form Cu2O segments by fragmenting the original CuO nanowires at the certain supersaturation density of oxygen vacancies. For unsheathed CuO nanowires, the reduction starts from the outer surface, which results in the nucleation and growth of Cu2O islands on the nanowire without generating significant oxygen vacancies in the bulk, which allows the unreacted zone of the CuO nanowire to survive at a higher temperature than the carbon-sheathed CuO nanowires. The formation of a coating shell on nanostructures can usually improve the stability of the nanostructures under harsh environments such as corrosive or electrochemical reactions.51–54 As shown here, such an effect may not be the case for oxides under high temperature environments. While the carbon shell suppresses the formation of Cu2O islands on the surface, the surface confinement by the coating layer promotes the generation of vacancies in the bulk that leads to the structure collapse at a lower temperature.
Since both the carbon-sheathed and unsheathed CuO nanowires were placed on a lacey amorphous carbon film for the in situ TEM heating experiments, possible local heating effect from the carbon coating layer can be minimized. As seen in Fig. 2(a and b), both the sheathed and unsheathed CuO nanowires are reduced to Cu2O under the same condition, suggesting that the local heat trap effect by the carbon coating is negligible. However, the reduction morphology of the carbon-sheathed portion is dramatically different from the unsheathed portion although the unsheathed CuO nanowires were also in direct contact with the carbon film (i.e. Fig. 3). This suggests that the chemical effect of the carbon sheath on the oxide reduction is negligible as well. As seen from Fig. 1 and 3, the temperature ramp rate for the reduction of carbon-sheathed CuO is ∼6.0 °C min−1, which is close to the ramp rate of 5.7 °C min−1 for the reduction of unsheathed CuO nanowires. Particularly, the samples shown in Fig. 1 and 2 were reduced under the same heating condition, the different reaction morphologies of the carbon-sheathed and unsheathed portions are related to the surface confinement effect.
To further elucidate if the coating carbon layer has had any chemical effect, we compared the reduction of carbon-sheathed and Al2O3-sheathed CuO nanowires. By employing ALD, we deposited a uniform ∼5 nm-thick coating layer of amorphous Al2O3 on CuO nanowires (as shown in Fig. 4(a and b)). The Al2O3-sheathed CuO nanowires were then reduced under vacuum at 450 °C for 1 h, which is similar as the reduction condition of carbon-sheathed and unsheathed CuO nanowires. Fig. 4(c) is a TEM image of the typical reduction morphology of the Al2O3-sheathed CuO nanowires, which reveals that the reduced CuO nanowire was fragmented into short segments. Fig. 4(d) is an SAED pattern from the segment shown in Fig. 4(c), which can be indexed well with Cu2O. These TEM results show that there are no significant differences in the reaction morphologies and reaction products for the reduction of both the carbon-sheathed and Al2O3-sheathed CuO nanowires, further indicating that the surface coating layer has little chemical effect on the oxide reduction process.
 | ||
| Fig. 4 (a) TEM image of a typical CuO nanowire coated with an amorphous Al2O3 layer by atomic layer deposition; (b) EDS analysis of the nanowire shown in (a) confirms the presence of Al (from the Al2O3 coating layer) and Cu from the CuO nanowire; (c) typical morphology of the Al2O3-coated CuO nanowires reduced at 450 °C for 1 h; (d) SAED pattern from the circled region in (c) demonstrates that the reduced CuO nanowire is fragmented to Cu2O segments. | ||
 | ||
| Fig. 5 Simulated CuO structures with the fixed (111) base: (a) a slab with stoichiometric O-terminated surfaces; (b) a single Cu2O layer at the surface of CuO; (c) a single Cu2O layer in the bulk of CuO. The surface and bulk simulations with (1 × 1 × 6) (111) CuO supercells were performed in the slab and periodic setups, respectively. For (b) and (c), the shown Cu2O and adjacent layers are doubled in the lateral directions to illustrate the VO ordering pattern. The small black spheres are O vacancies, the medium red spheres are O atoms, and the large grey spheres are Cu atoms. The shades of grey illustrate the coordination of the Cu atoms: from 1-fold (lightest) to 4-fold (darkest). | ||
Our in situ TEM observations (as shown in Fig. 3) demonstrate that the reduction of unsheathed CuO nanowires occurs via the nucleation and growth of Cu2O islands on the parent CuO nanowires by following the nucleation and growth model of oxide reduction. This is different from the “interfacial model” of oxide reduction for which the entire surface of the parent oxide is covered with a thin layer of the lower oxide very soon after the reduction reaction and the reaction boundary advances inward uniformly as the reaction proceeds (note that the “nucleation and growth model” also involves inward motion of the reaction boundary of individual nuclei and the reaction interface increases until growing nuclei overlap). However, for carbon-sheathed CuO nanowires, the reduction does not follow either the “nucleation and growth” or the “interface model” model. As revealed from our in situ TEM observations (i.e., Fig. 1), the reduction of carbon-sheathed CuO nanowires occurs via abrupt fragmentation into Cu2O segments without involving either the nucleation and growth of Cu2O islands or forming Cu2O/CuO interface.
To identify the microscopic origin of the surface confinement effect on the oxide reduction, we employ DFT + U to compare the energetics of CuO reduction at surface and in the bulk. We began our investigation of the CuO reduction energetics with the calculation of a single oxygen vacancy (VO) in a fixed 2 × 2 × 2 mS8-CuO supercell (the Pearson notation “mS8” indicates that the structure is monoclinic, side-centered, and has 8 atoms in the conventional unit cell). Referenced to molecular oxygen at T = 0 K, the formation energy was found to be EbulkVO = 3.90 eV/VO. The formation energy per VO remained essentially the same when two oxygen vacancies, furthest apart, were created in this 2 × 2 × 2 supercell. The apparently short range of the VO–VO interactions allowed us to use the minimum (2 × 1)-(111) base in the study of the single VO formation energy as a function of the distance to the surface in slab calculations with 6 unit cells (48 atoms). As shown in Fig. 5(a), the surface layer has four O and four Cu atoms, half of which are 3-fold coordinated and the other half are 4-fold coordinated for each species. We found that the formation energy decreased noticeably, by 0.79 eV/VO, only for the third top-most oxygen atom in the subsurface layer. For all other O atoms EVO showed smaller variations, below ∼0.2 eV/VO, from the bulk value of 3.90 eV/VO.
The effect of VO ordering at the CuO surface was examined by removing the top-most O atom and one of nearby O atoms. Because of the small size of the (2 × 1)-(111) base, creation of a second VO leads not to an isolated VO-VO pair but rather to an ordered O-depleted layer. For this reason the change in formation energy per VO is given below as an average for the two VO. A second vacancy created at the second top-most O site in the surface layer was found to have a substantially reduced  (by 0.99 eV/VO) compared to
(by 0.99 eV/VO) compared to  . With this particular VO arrangement shown in Fig. 5(b), the surface layer acquires the Cu2O stoichiometry and the four Cu atoms become 1-, 2-, 2-, and 3-fold coordinated. The configuration appears to be favorable because the Cu–O bonds for the 2-fold coordinated Cu atoms are at nearly 180 degrees, just as in the bulk cP6-Cu2O phase. In the two considerably less stable configurations of VO in the Cu2O surface layer, all Cu atoms are 2-fold coordinated but some of the Cu–O bonds are at 90 degrees. The observed stabilization of ordered VO is consistent with the key conclusions of Maimaiti et al.42 on the favorability of the CuO surface reduction to Cu2O.
. With this particular VO arrangement shown in Fig. 5(b), the surface layer acquires the Cu2O stoichiometry and the four Cu atoms become 1-, 2-, 2-, and 3-fold coordinated. The configuration appears to be favorable because the Cu–O bonds for the 2-fold coordinated Cu atoms are at nearly 180 degrees, just as in the bulk cP6-Cu2O phase. In the two considerably less stable configurations of VO in the Cu2O surface layer, all Cu atoms are 2-fold coordinated but some of the Cu–O bonds are at 90 degrees. The observed stabilization of ordered VO is consistent with the key conclusions of Maimaiti et al.42 on the favorability of the CuO surface reduction to Cu2O.
The influence of the surface on the reduction energetics was investigated further by moving the Cu2O layer just below the surface and then into the bulk. Slab and periodic setups with fixed (1 × 1 × 6) supercells with the (2 × 1)-(111) base were used in these simulations, respectively. As shown in Fig. 5(c), the reduced layer in these cases has three 2-fold coordinated and one 4-fold coordinated Cu atoms while the adjacent layers have two 3-fold coordinated Cu atoms which results in  and
and  . Creation of oxygen vacancies in the bulk is expected to induce local stress due to the slightly smaller measured volume of Cu4O2 (77.8 Å3)55 compared to that of Cu4O4 (81.0 Å3).56 Our DFT + U calculations at T = 0 K showed a comparable volume ratio of 78.9 Å3/83.3 Å3 for the two phases. To isolate the effect of the structural constraint we repeated the bulk calculations allowing the supercell to relax fully and observed only a small change from – 0.96 to −1.00 eV/VO in the relative formation energy of the Cu2O layer. Comparison of these values to the one obtained for the surface,
. Creation of oxygen vacancies in the bulk is expected to induce local stress due to the slightly smaller measured volume of Cu4O2 (77.8 Å3)55 compared to that of Cu4O4 (81.0 Å3).56 Our DFT + U calculations at T = 0 K showed a comparable volume ratio of 78.9 Å3/83.3 Å3 for the two phases. To isolate the effect of the structural constraint we repeated the bulk calculations allowing the supercell to relax fully and observed only a small change from – 0.96 to −1.00 eV/VO in the relative formation energy of the Cu2O layer. Comparison of these values to the one obtained for the surface,  , reveals that the CuO reduction proceeds more easily for layers (i) with the starting 4-fold coordination of Cu and O atoms and (ii) near the surface where the O-depleted configurations are able to relieve stress.
, reveals that the CuO reduction proceeds more easily for layers (i) with the starting 4-fold coordination of Cu and O atoms and (ii) near the surface where the O-depleted configurations are able to relieve stress.
These DFT calculations corroborate well with our experimental observations on the unsheathed CuO nanowires for which the surface reduction is more favorable than the bulk reduction because of the smaller oxygen vacancy formation energy. The previously investigated ordering of surface oxygen vacancies resulting in Cu2O formation near the CuO surface42 is consistent with the experimentally observed Cu2O nucleation and growth on the nanowire surface. The illustrated strong tendency of VO to order in the bulk as well as at the surface suggests that Cu2O nucleation may occur in carbon-sheathed CuO nanowires with the presence of substantial amounts of bulk VO.
4. Conclusion
In summary, we performed a comparative in situ heating TEM study of the thermally induced reduction of CuO nanowires with and without a carbon shell. We find that carbon-sheathed CuO nanowires are reduced to Cu2O by the fragmentation of the starting CuO nanowire into Cu2O segments encapsulated by the carbon shell, while unsheathed CuO nanowires are reduced to Cu2O with the nucleation and growth of Cu2O bulges on the nanowire surface. We show that their difference originates from the carbon shell surface confinement effect that changes the reaction mechanism from surface reduction via nucleation and growth of 3D Cu2O islands on the nanowire surface to the internal reduction. One of the possible reduction mechanisms in the bulk, corroborated with our DFT calculations, is via ordering of oxygen vacancies in the bulk which can lead to nanowire fragmentation into CuO segments.Acknowledgements
This work was supported by the National Science Foundation under NSF CAREER Award Grant CMMI-1056611.References
- V. E. Henrich and P. A. Cox, The surface science of metal oxides. Cambridge University Press, Cambridge, 1994 Search PubMed.
- H. H. Kung, Transition metal oxides: surface chemistry and catalysis, Elsevier, New York, 1989 Search PubMed.
- C. H. Bamford, C. F. H. Tipper, R. G. Compton, Comprehensive Chemical Kinetics, Elsevier, New York, 1984, vol. 21 Search PubMed.
- S. R. Zhang, J. J. Shan, Y. Zhu, L. Nguyen, W. X. Huang, H. Yoshida, S. Takeda and F. Tao, Nano Lett., 2013, 13, 3310–3314 CrossRef CAS PubMed.
- R. D. L. Smith, M. S. Prevot, R. D. Fagan, S. Trudel and C. P. Berlinguette, J. Am. Chem. Soc., 2013, 135(31), 11580–11586 CrossRef CAS PubMed.
- S. D. Senanayake, D. Stacchiola and J. A. Rodriguez, Acc. Chem. Res., 2013, 46(8), 1702–1711 CrossRef CAS PubMed.
- D. Gamarra, A. Lopez Camara, M. Monte, S. B. Rasmussen, L. E. Chinchilla, A. B. Hungria, G. Munuera, N. Gyorffy, Z. Schay, V. C. Corberan, J. C. Conesa and A. Martinez-Arias, Appl. Catal., B, 2013, 130–131, 224–238 CrossRef CAS PubMed.
- K. Morita, K. Sakuma, K. Miyajima and F. Mafune, J. Phys. Chem. A, 2013, 117(40), 10145–10150 CrossRef CAS PubMed.
- R. Baghi, G. R. Peterson and L. J. Hope-Weeks, J. Mater. Chem. A, 2013, 1, 10898–10902 CAS.
- Y. Z. Hu, R. Sharangpani and S. P. Tay, J. Electrochem. Soc., 2001, 148(12), G669–G675 CrossRef CAS PubMed.
- S. Y. Lee, N. Mettlach, N. Nguyen, Y. M. Sun and J. M. White, Appl. Surf. Sci., 2003, 206, 102–109 CrossRef CAS.
- R. Govindaraj, C. S. Sundar and R. Kesavamoorthy, J. Appl. Phys., 2006, 100, 084318 CrossRef PubMed.
- F. Irrera, G. Puzzilli and D. Caputo, Microelectron. Reliab., 2005, 45, 853–856 CrossRef CAS PubMed.
- J. Li, J. Mayer and K. Tu, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45(10), 5683–5686 CrossRef CAS.
- J. Y. Kim, J. A. Rodriguez, J. C. Hanson, A. I. Frenkel and P. L. Lee, J. Am. Chem. Soc., 2003, 125, 10684–10692 CrossRef CAS PubMed.
- J. A. Rodriguez, J. C. Hanson, A. I. Frenkel, J. Y. Kim and M. Pérez, J. Am. Chem. Soc., 2002, 124(2), 346–354 CrossRef CAS PubMed.
- J. J. Scholz and M. A. Langell, Surf. Sci., 1985, 164(2–3), 543–557 CrossRef CAS.
- R. Furstenau, G. McDougall and M. Langell, Surf. Sci., 1985, 150(1), 55–79 CrossRef CAS.
- B. Delmon, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knozinger andJ. Weitkamp, Wiley-VCH, New York, 1997, pp. 264–277 Search PubMed.
- J. A. Rodriguez, J. Y. Kim, J. C. Hanson, M. Perez and A. I. Frenkel, Catal. Lett., 2003, 85(3–4), 247–254 CrossRef CAS.
- X. Q. Wang, J. C. Hanson, A. I. Frenkel, J. Y. Kim and J. A. Rodrigues, J. Phys. Chem. B, 2004, 108, 13667–13673 CrossRef CAS.
- T. Ressler, R. E. Jentoft, J. Wienold, M. M. Gunter and O. Timpe, J. Phys. Chem. B, 2000, 104, 6360–6370 CrossRef CAS.
- J. Pike, S. W. Chan, F. Zhang, X. Q. Wang and J. Hanson, Appl. Catal., A, 2006, 303, 273–277 CrossRef CAS PubMed.
- G. W. Zhou and J. C. Yang, Phys. Rev. Lett., 2004, 93, 226101 CrossRef.
- G. W. Zhou, W. Y. Dai and J. C. Yang, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77(24), 245427 CrossRef.
- Y. Qin, S. M. Lee, A. L. Pan, U. Gosele and M. Knez, Nano Lett., 2008, 8(1), 114–118 CrossRef CAS PubMed.
- L. Li and G. W. Zhou, Surf. Sci., 2011, 605, 54–61 CrossRef CAS PubMed.
- L. Yuan, Q. Yin, Y. Wang and G. Zhou, Chem. Phys. Lett., 2013, 590, 92–96 CrossRef CAS PubMed.
- Y. Li, Q. Fu and M. Flytzani-Stephanopoulos, Appl. Catal., B, 2000, 27(3), 179–191 CrossRef CAS.
- C. Ammon, A. Bayer, G. Held, B. Richer, L. Schmidt and H. Steinruck, Surf. Sci., 2002, 507, 845–850 CrossRef.
- C. T. Campbell and K. A. Daube, J. Catal., 1987, 104(1), 109–119 CrossRef CAS.
- T. Tabakova, V. Idakiev, J. Papavasiliou, G. Avgouropoulos and T. Loannides, Catal. Commun., 2007, 8(1), 101–106 CrossRef CAS PubMed.
- I. Nakamura, H. Nakano, T. Fujitani, T. Uchijima and J. Nakamura, J. Vac. Sci. Technol., A, 1999, 17(4), 1592–1595 CAS.
- X. Q. Wang, J. A. Rodriguez, J. C. Hanson, D. Gamarra, A. Martinez-Arias and M. Fernandez-Garcia, J. Phys. Chem. B, 2005, 109(42), 19595–19603 CrossRef CAS PubMed.
- L. Yuan and G. W. Zhou, J. Electrochem. Soc., 2012, 159, C205–C209 CrossRef CAS PubMed.
- R. Mema, L. Yuan, Q. Du, Y. Wang and G. W. Zhou, Chem. Phys. Lett., 2011, 512(1–3), 87–91 CrossRef CAS PubMed.
- L. Yuan, Y. Q. Wang, R. Mema and G. W. Zhou, Acta Mater., 2011, 59, 2491–2500 CrossRef CAS PubMed.
- W. Ding, D. Dikin, X. Chen, R. Piner, R. Ruoff, E. Zussman, X. Wang and X. Li, J. Appl. Phys., 2005, 98(1), 014905 CrossRef PubMed.
- E. Sutter, P. Sutter and Y. Zhu, Nano Lett., 2005, 5(10), 2092–2096 CrossRef CAS PubMed.
- V. C. Holmberg, M. G. Panthani and B. A. Korgel, Science, 2009, 326(5951), 405–407 CrossRef CAS PubMed.
- L. Y. Isseroff and E. A. Carter, Chem. Mater., 2013, 25(3), 253–265 CrossRef CAS.
- Y. Maimaiti, M. Nolan and S. D. Elliott, Phys. Chem. Chem. Phys., 2014, 16, 3036–3046 RSC.
- V. I. Anisimov, J. Zaanen and O. K. Andersen, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44(3), 943 CrossRef CAS.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2006, 124(21), 9906 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77(18), 3865–3868 CrossRef CAS.
- D. Wu, Q. Zhang and M. Tao, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73(23), 235206 CrossRef.
- M. Heinemann, B. Eifert and C. Heiliger, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87(11), 115111 CrossRef.
- J. Hu, D. Li, J. G. Lu and R. Wu, J. Phys. Chem. C, 2010, 114(40), 17120–17126 CAS.
- M.-F. Yu, O. Lourie, M. J. Dyer, K. Moloni, T. F. Kelly and R. S. Ruoff, Science, 2000, 287(5453), 637–640 CrossRef CAS.
- D. N. Madsen, K. Mølhave, R. Mateiu, A. M. Rasmussen, M. Brorson, C. J. Jacobsen and P. Bøggild, Nano Lett., 2003, 3(1), 47–49 CrossRef CAS.
- A. Martinez-Garcia, V. K. Vendra, S. Sunkara, P. Haldankar, J. Jasinski and M. K. Sunkara, J. Mater. Chem. A, 2013, 1, 15235–15241 CAS.
- L. F. Shen, H. S. Li, E. Uchaker, X. G. Zhang and G. Z. Cai, Nano Lett., 2012, 12(11), 5673–5678 CrossRef CAS PubMed.
- C. Marichy, M. Bechelany and N. Pinna, Adv. Mater., 2012, 24, 1017–1032 CrossRef CAS PubMed.
- S. Carenco, C. Surcin, M. Morcrette, D. Larcher, N. Mezailles, C. Boissiere and C. Sanchez, Chem. Mater., 2012, 24(4), 688–697 CrossRef CAS.
- S. Hafner and S. Nagel, Phys. Chem. Miner., 1983, 9(1), 19–22 CrossRef CAS.
- S. Asbrink and L.-J. Norrby, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1970, 26(1), 8–15 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |