
Hyperbranched polyethers with tunable glass transition temperature: controlled synthesis and mixing rules†
Abstract
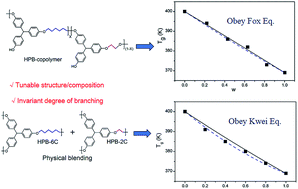
By taking advantage of competing side reactions, controlled synthesis of a series of homo- and co-polymerized hyperbranched polyethers (HBPEs) is demonstrated using AB2 monomers of different spacer lengths. This reacting system shows good controllability and scalability. More importantly, the degree of branching is found to be insensitive to the molecular weight and spacer length in monomers. Thus, the value and width of Tg can be tuned by varying monomer spacer length, terminal groups, molecular weight, as well as by copolymerization and physical blending. The dependence of Tg in binary homopolymer blends on composition and the dependence of Tg in copolymers on monomer ratio are established and compared for the first time. Tg of copolymers obeys the Fox equation, whereas Tg in binary blends only follows the Kwei equation. Copolymerization does not increase the width of Tg. In contrast, the width of Tg of binary blends is much broader than that of copolymers, even though the broadening in Tg can be reduced by increasing the polarity of terminal groups.
 Please wait while we load your content...
Please wait while we load your content...

