
Reactivity of nitric oxide with ruthenium complexes derived from bidentate ligands: structure of a ruthenium nitrosyl complex, photoinduced generation and estimation of nitric oxide†
Abstract
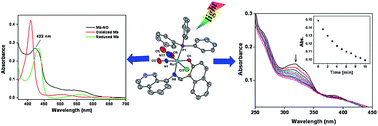
Ruthenium complexes [Ru(L1)(PPh3)2(CO)Cl] (1) [where L1H is (E)-2-((naphthalen-1-ylimino)methyl)phenol] and [Ru(L2)(PPh3)2(CO)Cl] (2) [where L2H is (E)-1-((naphthalen-1-ylimino) methyl) naphthalene-2-ol] [H stands for dissociable proton] were synthesized and characterized by UV-Vis, IR and NMR spectral studies. Nitric oxide reactivity studies afforded ruthenium nitrosyl complexes [Ru(L1)(PPh3)(NO2)(NO)Cl] (1a) and [Ru(L2)(PPh3)(NO2)(NO)Cl] (2a) from complexes 1 and 2, respectively. Complexes 1a and 2a were characterized by different spectroscopic studies. The molecular structure of complex 2a was determined by X-ray crystallography. The redox properties of all the complexes were investigated by cyclic voltammetry. Coordinated nitric oxide was found to be photolabile and photogenerated NO was trapped by reduced myoglobin and liberated NO was estimated by the Griess reaction.
 Please wait while we load your content...
Please wait while we load your content...

