 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecularly imprinted polymer beads for nicotine recognition prepared by RAFT precipitation polymerization: a step forward towards multi-functionalities†
Tongchang Zhouab,
Lars Jørgensenc,
Maria Ahlm Mattebjergc,
Ioannis S. Chronakisc and
Lei Ye*a
aDivision of Pure and Applied Biochemistry, Lund University, Box 124, 22100 Lund, Sweden. E-mail: lei.ye@tbiokem.lth.se; Fax: +46 46 2224611; Tel: +46 46 2229560
bGuangzhou Pharmaceutical Holdings, Limited, Guangzhou 510130, China
cDTU Food, Technical University of Denmark, Søltofts Plads B227, DK-2800 Kgs, Lyngby, Denmark
First published on 30th June 2014
Abstract
A nicotine imprinted polymer was synthesized by reversible addition-fragmentation chain transfer (RAFT) polymerization using methacrylic acid (MAA) as a functional monomer. The resulting molecularly imprinted polymers were monodispersed beads with an average diameter of 1.55 μm. The molecular selectivity of the imprinted polymer beads was evaluated by studying the uptake of nicotine and its structural analogs by the polymer beads. Equilibrium binding results indicate that the amount of nicotine bound to the imprinted polymer beads is significantly higher than that bound to the non-imprinted polymer in both acetonitrile and in a mixture of acetonitrile and water. The RAFT reagent present on the surface of the polymer beads allowed straightforward grafting of hydrophilic polymer brushes on the particle surface. In addition to the demonstrated molecular selectivity and the straightforward surface modification of the imprinted polymer beads, we also show that the dithioester end groups on the surface of the polymer beads can be converted into new thiol groups without sacrificing the specific molecular recognition. Through the new terminal thiol groups, a fluorescent dye was conveniently conjugated to the imprinted polymer beads via Michael addition reaction. The living characteristic of RAFT and the versatile thiol groups that can be derived from the RAFT reagent provide many new possibilities for realizing multi-functionalities for molecularly imprinted polymers.
Introduction
Molecularly imprinted polymers (MIPs) have been used for selective binding in many fields due to their capability of specific molecular recognition.1–3 Currently, MIPs are being developed for various applications such as in chemical sensors, drug delivery, protein crystallization, and so on.4–6 MIPs can be prepared in a variety of physical forms by using different polymerization techniques. The traditional methods such as bulk polymerization7 and suspension polymerization8 have been used frequently to prepare MIPs since these methods are simple and efficient on a laboratory scale. However, MIPs prepared by these traditional methods are irregular in size and shape, and can only be obtained on a small scale. In order to overcome these limitations, many new synthetic methods have been developed to prepare monodispersed MIP particles.9,10 Often, spherical polymer beads can be obtained in good yields using these methods, especially the precipitation polymerization method that does not require interfering reagents in the reaction system.Among the recently developed methods to produce monodispersed MIP microspheres, precipitation polymerization is the most popular due to its simple implementation in standard chemical laboratories. More sophisticated polymerization techniques, such as controlled/living radical polymerization (CRP),11–14 have drawn great interest for preparing MIP microspheres, because the CRP techniques allow more precise control over the molecular weight, composition and end group functionality of the obtained polymers. Among the different CRP techniques that are available, reversible addition-fragmentation chain transfer (RAFT) polymerization is particularly attractive, because it is compatible with a wide range of functional monomers and does not involve the use of special catalyst.15–18 Zhang and co-workers have carried out a series of RAFT precipitation polymerization to prepare uniform MIP microspheres,19,20 and demonstrated that by grafting a hydrophilic polymer layer using the living character of RAFT, the imprinted microspheres can be turned into water-compatible, i.e. the modified MIP microspheres displayed much reduced non-specific binding in aqueous solution.13,21
In previous studies, we have shown that uniform MIP nanoparticles and microspheres can be used as versatile building blocks to assemble multi-functional composites and hierarchical structures.22–25 To enable MIP nanoparticles and microspheres to act as useful building blocks, it is necessary to introduce new functional groups on the particle surface without affecting the specific molecular recognition in the core of the particles. In this respect, the living character of RAFT and the possibility of converting surface bound RAFT agent into new functional groups have drawn our attention, as these chemical reactions are non-destructive for the imprinted binding sites in the interior of the MIP particles, and the reactions can be carried out under relatively mild conditions. Although surface modification of MIP microspheres has been achieved using RAFT-mediated grafting polymerization, no research has been reported on surface modification of MIPs by converting RAFT agent directly into new functional groups. In this work we demonstrate, for the first time, that by converting surface accessible RAFT agent into thiol groups, new functionalities (e.g. fluorescence) can be introduced to MIP microspheres in a straightforward manner. As a model, we prepared nicotine imprinted polymer beads using RAFT precipitation polymerization. The living character of the MIP microspheres was first verified by on-particle synthesis of a temperature-responsive poly(N-isopropylacrylamide) using RAFT-mediated grafting polymerization. By converting the RAFT agent into terminal thiol groups, we also show that new fluorescent dyes can be conjugated to the MIP particles through a Michael addition reaction.
Nicotine is a potent parasympathomimetic alkaloid that is widely used as a stimulant drug in smoking cession therapy.26–28 To achieve an ideal therapeutic delivery for the purpose of smoking intervention, nicotine has been introduced into chewing gums and transdermal patches to realize its controlled release.29 In the literature, MIPs have been suggested as potential drug delivery materials based on their capability of retaining and controllable release of therapeutic compounds.30 In this paper we present, for the first time, the synthesis of nicotine imprinted uniform polymer microspheres using RAFT precipitation polymerization, and the use of the RAFT agent to achieve straightforward surface modification towards multi-functional materials with pre-designed molecular recognition properties. Besides potential applications as building block materials (e.g. to prepare nicotine patches), the new MIP beads may also be used as a basic component to develop chemical sensors for monitoring nicotine concentration.
Experimental
Materials
Nicotine, cotinine, myosmine, N-isopropylacrylamide, ethylene glycol dimethacrylate (EGDMA, 98%), N-(1-pyrenyl)maleimide (NPM), N,N-dimethylformamide (DMF) and cumyl dithiobenzoate (CDB, 99%) were purchased from Sigma-Aldrich. Methacrylic acid (MAA, 98.5%) and azobisisobutyronitrile (AIBN, 98%) were purchased from Merck (Darmstadt, Germany). L-[N-methyl-3H]-nicotine (specific activity = 80.4 Ci per mmol, 12.4 μM in ethanol) was obtained from NEN Life Science Products, Inc. (Boston, MA). Scintillation liquid (Ecoscint A) was from National Diagnostics (Atlanta, GA). AIBN was recrystallized from methanol before use. Other chemicals were analytical grade and were used as received from commercial suppliers. All the aqueous solutions were prepared in ultra-pure water (18 MΩ cm).Synthesis of molecularly imprinted polymer
Nicotine imprinted polymer microspheres were synthesized by RAFT precipitation polymerization in acetonitrile. Briefly, nicotine (80 μL, 0.5 mmol), MAA (111 μL, 1.3 mmol), EGDMA (1200 μL, 6.4 mmol), AIBN (25 mg) and CDB (60 mg) were dissolved in 40 mL acetonitrile. The solution was added into a one-neck round bottom flask (100 mL) and purged with nitrogen for 30 minutes. The flask was then attached to the rotor-arm of a rotary evaporator. The polymerization was initiated at 60 °C and continued for 24 h while the flask was rotated at a speed of 30 rpm.After the polymerization, the solid particles were collected by centrifugation and washed with methanol for two times. To remove the template, the microspheres were washed with methanol containing 10% acetic acid until no template could be detected from the washing solvent. Finally the polymer beads were washed with methanol for two times and acetonitrile for three times before dried in a vacuum chamber. The obtained polymer product was named as MIP.
As a control, non-imprinted polymer (NIP) microspheres were synthesized in the same way as the MIP, except that no template was added in the initial monomer solution.
On-particle synthesis of poly(N-isopropylacrylamide) by RAFT polymerization
To graft poly(N-isopropylacrylamide) brushes, the polymer beads obtained in the previous step (100 mg) were dispersed in 5 mL DMF. After addition of N-isopropylacrylamide (2 g), CDB (5 mg) and AIBN (1 mg), the particle suspension was purged with nitrogen for 10 minutes. The reaction mixture was then heated to 70 °C and kept for 24 h under vigorous stirring. After the polymerization, the particles were collected by centrifugation, washed with methanol and then dried in a vacuum chamber.Converting RAFT agent into terminal thiols by aminolysis
The dithioester end groups on the MIP and NIP particles were converted into terminal thiols by treatment with organic amines.31 The polymer microspheres (MIP or NIP, 200 mg), ethylene diamine (110 μL) and triethylamine (230 μL) were mixed in DMF (3.3 mL) in a 10 mL one-neck round bottom flask. After being purged with nitrogen for 5 minutes, the reaction mixture was stirred at 50 °C for 16 h. After the aminolysis reaction, the particle suspension was acidified to ∼pH 5 by adding 0.1 M HCl. The particles were finally washed with water until the pH of the supernatant became neutral. In this way the dithioester groups on the surface of particles were transformed into thiol groups. These modified polymer particles (now covered by terminal thiol groups) derived from the MIP and NIP microspheres were named as MIP-SH and NIP-SH, respectively.Conjugation of thiol-terminated particles with fluorescent dye
NPM (3 mg) was dissolved in 10 mL of acetonitrile. The NPM solution (750 μL) was mixed with 10 mM Tris buffer (750 μL, pH 9) before the thiol-terminated particles (100 mg) were added. The particle suspension was gently stirred on a rocking table for 1 h. After this treatment, the particles were separated by centrifugation, washed thoroughly with water and dried in a vacuum chamber.Determination of thiol groups in MIP-SH
The number of thiol groups in MIP-SH particles was determined using the thiol-specific reagent NPM. MIP-SH particles (10 mg) were added into a mixture of 10 mM Tris buffer (500 μL, pH 9) and NPM solution (500 μL, 6 μM). The mixture was gently stirred at room temperature for 1 h. After centrifugation, about 600 μL of supernatant was collected. The concentration of NPM in the supernatant was measured with a DU800 Spectrophotometer (Beckman Coulter) at 339 nm.Characterization
Attenuated total reflection (ATR) FTIR analysis was carried out on a Nicolet iS5 FT-IR Spectrophotometer (Thermo Scientific). The dry particles were transferred onto the sample plate of the FTIR instrument and analysed directly. All spectra were collected at room temperature in the 4000 cm−1 to 500 cm−1 region using 16 scans.Scanning electron microscopy analysis was carried out on a FEI Quanta 200 ESEM FEG scanning electron microscope. The polymer particles were sputter coated with gold at 40 mA for 10 seconds before the SEM analysis.
Fluorescence spectra were collected using a QuantaMaster C-60/2000 spectrofluorometer (Photon Technology International, Lawrenceville, NJ, USA).
Equilibrium binding analysis
Polymer particles were mixed with different test compounds in 1 mL solvent (acetonitrile, water or solvent mixture). The mixture was gently stirred on a rocking table at room temperature for 16 h. After centrifugation, supernatant (600 μL) from each sample was collected, in which the concentration of the test compound was measured using a DU800 UV-Vis Spectrophotometer (Beckman Coulter). The wavelength used in the UV detection was fixed at 260 nm for nicotine, 255 nm for cotinine and 234 nm for myosmine, respectively. The amount of the bound compound was calculated as the difference between the total and the free compound.Radioligand binding analysis
Various amounts of polymer particles were mixed with 1 mL of acetonitrile containing L-[N-methyl-3H]-nicotine (223 fmol) in a series of micro centrifuge tubes. The mixture was incubated on a rocking table at room temperature overnight. After the incubation, the polymers were sedimented by centrifugation at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min. Supernatant (500 μL) was withdrawn and mixed with 10 mL of scintillation liquid (Ecoscint A). The radioactivity of the samples was measured with a Tri-Carb 2800TR liquid scintillation analyzer (PerkinElmer). The amount of radioligand bound to the polymer particles was calculated by subtracting the free radioligand from the total radioligand added. The data are mean values of measurements on three independent samples.
000 rpm for 10 min. Supernatant (500 μL) was withdrawn and mixed with 10 mL of scintillation liquid (Ecoscint A). The radioactivity of the samples was measured with a Tri-Carb 2800TR liquid scintillation analyzer (PerkinElmer). The amount of radioligand bound to the polymer particles was calculated by subtracting the free radioligand from the total radioligand added. The data are mean values of measurements on three independent samples.
In competitive radioligand binding experiments, the imprinted particles (0.05 mg) were mixed with L-[N-methyl-3H]-nicotine (223 fmol) and different amount of nicotine or cotinine in 1 mL of acetonitrile. The amount of radioligand bound to the polymer particles was calculated by subtracting the free radioligand from the total radioligand added.
Results and discussion
Uniform nicotine-imprinted polymer beads obtained by RAFT precipitation polymerization
In previous studies, we have shown that well-defined molecularly imprinted polymer beads can be prepared by precipitation polymerization under optimized solvent conditions.10 Compared to uncontrolled radical polymerization, Zhang and co-workers have shown that by using controlled (living) radical polymerization technique, precipitation polymerization can lead to nearly monodisperse MIP microspheres with well retained molecular recognition properties. One important advantage of combining CRP with precipitation polymerization is that it gives uniform MIP microspheres covered with “living” end groups, which can be utilized to graft a new layer of polymer for straightforward surface functionalization.13,21 Besides, it has been suggested that the unique characteristics of CRPs, i.e. the rapid initiation, slow chain propagation and lack of early termination, make the obtained polymer microspheres to have a more homogeneous network structure as compared to the traditional polymer beads synthesized using uncontrolled radical polymerization.20 Based on these attractive features of CRP and precipitation polymerization, in this work we decided to investigate the preparation of nicotine imprinted polymer microspheres that can be used in future drug delivery systems or as the basic molecular recognition components in nicotine sensors.The nicotine imprinted polymer particles synthesized by RAFT precipitation polymerization were first characterized by scanning electron microscopy. As seen in Fig. 1, the size of the MIP and the NIP microspheres are 1.55 ± 0.04 μm (Fig. 1a) and 4.33 ± 0.07 μm (Fig. 1b), respectively. We should note that when a traditional uncontrolled radical polymerization was used, the imprinting reaction resulted in significantly smaller particles, which were difficult to recover even with high speed centrifugation (data not shown). The larger particle size achieved by the RAFT precipitation polymerization is in agreement with that reported in the previous literature.13,21 Under the RAFT polymerization condition, the relatively fast initiation and the simultaneous growth of the living polymer radicals make it possible that particle nucleation takes place in the early reaction phase, and in the following phase polymer chains grow from all the existing particles. Because of the different particle nucleation and growth mechanism, RAFT precipitation polymerization produces more uniform MIP and NIP microspheres than the conventional radical precipitation polymerization.
 | ||
| Fig. 1 SEM images of polymer microspheres MIP (a), NIP (b), MIP-SH (c), and NIP-SH (d). | ||
Molecular selectivity of nicotine-imprinted polymer beads
To verify that the MIP beads obtained by the RAFT precipitation polymerization have the desired molecular selectivity, we first compared the equilibrium binding of nicotine with the MIP and the NIP microspheres in acetonitrile. As shown in Fig. 2, the uptake of nicotine by the MIP is significantly (3 to 4 times) higher than the NIP, implying that the MIP possesses a much higher affinity for nicotine due to the molecularly imprinted sites. The specific nicotine binding is supposed to be mediated by the interaction between the carboxyl groups in the MIP and the basic amine moiety in nicotine. | ||
| Fig. 2 Equilibrium binding of nicotine to the MIP (■) and the NIP (●) microspheres. The particle concentration was 5 mg mL−1 in acetonitrile. | ||
To study the selectivity of the imprinted polymer particles, we tested the binding of two structural analogs of nicotine with the MIP and NIP microspheres. As shown in Fig. 3, the binding of myosmine and cotinine to MIP was only around 10%, which is significantly lower than nicotine binding (50%). When tested on the non-imprinted polymer, both nicotine and its structural analogues show much lower binding. These results further confirm that the high nicotine binding by the MIP was contributed by the imprinted sites rather than by the random carboxyl groups found in the NIP microspheres.
 | ||
| Fig. 3 (a) Uptake of nicotine and its structural analogues (0.155 mM) by MIP and NIP microspheres (5 mg) in 1 mL of acetonitrile. (b) Chemical structures of the tested analytes. | ||
It is well known that solvent plays an important role in determining the MIP binding process because it affects the surface properties of MIP and the formation of selective H-bonds. Fig. 4 shows the effect of water on the binding of nicotine by the MIP and NIP microspheres. In pure water, uptake of nicotine by MIP was only slightly higher than NIP, and both MIP and NIP displayed high nicotine binding due to non-specific interactions. By increasing acetonitrile content, the non-specific binding was significantly reduced, resulting in the best specific binding of nicotine (40.5%) to occur in pure acetonitrile.
 | ||
| Fig. 4 Uptake of nicotine (0.155 mM) by 5 mg of polymer microspheres in water containing different amount of acetonitrile. | ||
Modification of MIP microspheres by surface RAFT polymerization
Surface modification of organic polymer particles can improve their compatibility with practical application environments and therefore has attracted increasing interest. The living character of RAFT polymerization products makes it possible to graft new layers of polymers by simply carrying out a second RAFT polymerization with new monomer feed. To demonstrate the living property of the imprinted polymer microspheres, we grafted poly(N-isopropylacrylamide) (polyNIPAm) brushes on the surface of the MIP. To suppress radical–radical coupling reactions on particle surface, some free RAFT agent was added in the reaction mixture to minimize chain termination.12,32 After the graft polymerization, we used ATR-FTIR to characterize the modified MIP particles (MIP–polyNIPAm). As shown in Fig. 5a, the IR band at 1654 cm−1 and 1541 cm−1 can be assigned to the fundamental stretching vibration of Amide I and Amide II, respectively, which are from the polyNIPAm brushes. These characteristic IR bands support that the RAFT reagent in the MIP microspheres indeed allow continuous grafting of new polymer chains for effective surface modification. | ||
| Fig. 5 FTIR spectra of (a) MIP–polyNIPAm, (b) MIP, and (c) MIP-SH. | ||
Introduction of thiol groups to MIP microspheres
As a very versatile functional group, terminal thiols can be utilized to conjugate different organic molecules through S–S bridges, or via simple Michael addition reaction or thiol-ene click reaction.33–37 The possibility of using terminal thiols to immobilize MIP particles on Au-coated transducers to develop different chemical sensors and to introduce additional functional properties (e.g. stimuli responsiveness) to MIPs is also appealing for practical applications of molecularly imprinted polymers.The covalently bonded RAFT agent (CDB) in the MIP and NIP particles was converted into thiol groups by treating the particles with organic amines.16 After this aminolysis reaction, the size of both the MIP and the NIP particles remained unchanged (Fig. 1c and d), although the colour of the particles changed from pink to white. In the ATR-FTIR analysis, the intensity of the IR band at 1048 cm−1 (corresponding to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S stretching) decreased (Fig. 5c), which suggests that part of the CDB moieties has been altered. To prove that the CDB moieties have been converted into terminal thiols, we labelled the MIP-SH particles with a thiol-selective, fluorogenic pyrene derivative NPM. As seen in Fig. 6, after mixing MIP-SH particles with NPM, the particle suspension became strongly fluorescent. As only the product of the Michael addition reaction between MIP-SH and NPM can be fluorescent, it is clear that the aminolysis reaction has turned the RAFT agent into thiol groups.
S stretching) decreased (Fig. 5c), which suggests that part of the CDB moieties has been altered. To prove that the CDB moieties have been converted into terminal thiols, we labelled the MIP-SH particles with a thiol-selective, fluorogenic pyrene derivative NPM. As seen in Fig. 6, after mixing MIP-SH particles with NPM, the particle suspension became strongly fluorescent. As only the product of the Michael addition reaction between MIP-SH and NPM can be fluorescent, it is clear that the aminolysis reaction has turned the RAFT agent into thiol groups.
 | ||
| Fig. 6 Fluorescence spectra of NPM solution (300 mg L−1) in acetonitrile: (a) after addition of MIP-SH (10 mg L−1, excitation wavelength 343 nm), (b) before addition of MIP-SH (excitation wavelength at 360 nm), (c) before addition of MIP-SH (excitation wavelength 343 nm). | ||
To quantify the amount of the thiol groups in MIP-SH particles, we first treated the particles with excess of NPM, and then measured the remaining NPM by UV spectroscopy. The density of thiol groups in MIP-SH was found to be 122 μmol g−1, which indicates that approximately 68% of the dithioester structure (CDB, based on theoretical value) has been changed into –SH groups.
Molecular recognition property of MIP-SH microspheres
To study the molecular recognition properties of the thiol-functionalized particles, we compared nicotine binding with the different polymer microspheres in acetonitrile. As shown in Fig. 7, the aminolysis treatment resulted in an obvious reduction of nicotine binding for the imprinted polymer (MIP-SH vs. MIP), despite that the specific binding of nicotine remains significant, i.e. MIP-SH binds 3 times more nicotine than NIP-SH. Considering that a significant amount of CDB moiety (68%) was converted into –SH groups during the aminolysis, we suggest that the part of the covalently bonded RAFT agent in the MIP microspheres also participated in the specific binding, presumably through π–π interaction with the aromatic ring of nicotine. | ||
| Fig. 7 Uptake of nicotine (0.155 mM) in acetonitrile by the imprinted and non-imprinted polymer microspheres (5 mg mL−1) before and after the aminolysis treatment. | ||
To investigate more in detail how the aminolysis affects the molecular recognition sites in the imprinted particles, we carried out radioligand binding analysis for nicotine, because the high sensitivity of this technique allows us to gain more insights into the high affinity sites. Fig. 8a shows clearly that when very low concentration of nicotine was used, the thiol-functionalized MIP-SH still displayed significantly high nicotine binding than NIP-SH. We should note that at a polymer concentration of 0.05 mg mL−1, MIP-SH could bind ∼40% of the labelled nicotine, which is somewhat lower than the uptake of the radioligand by the untreated MIP particles (63%).
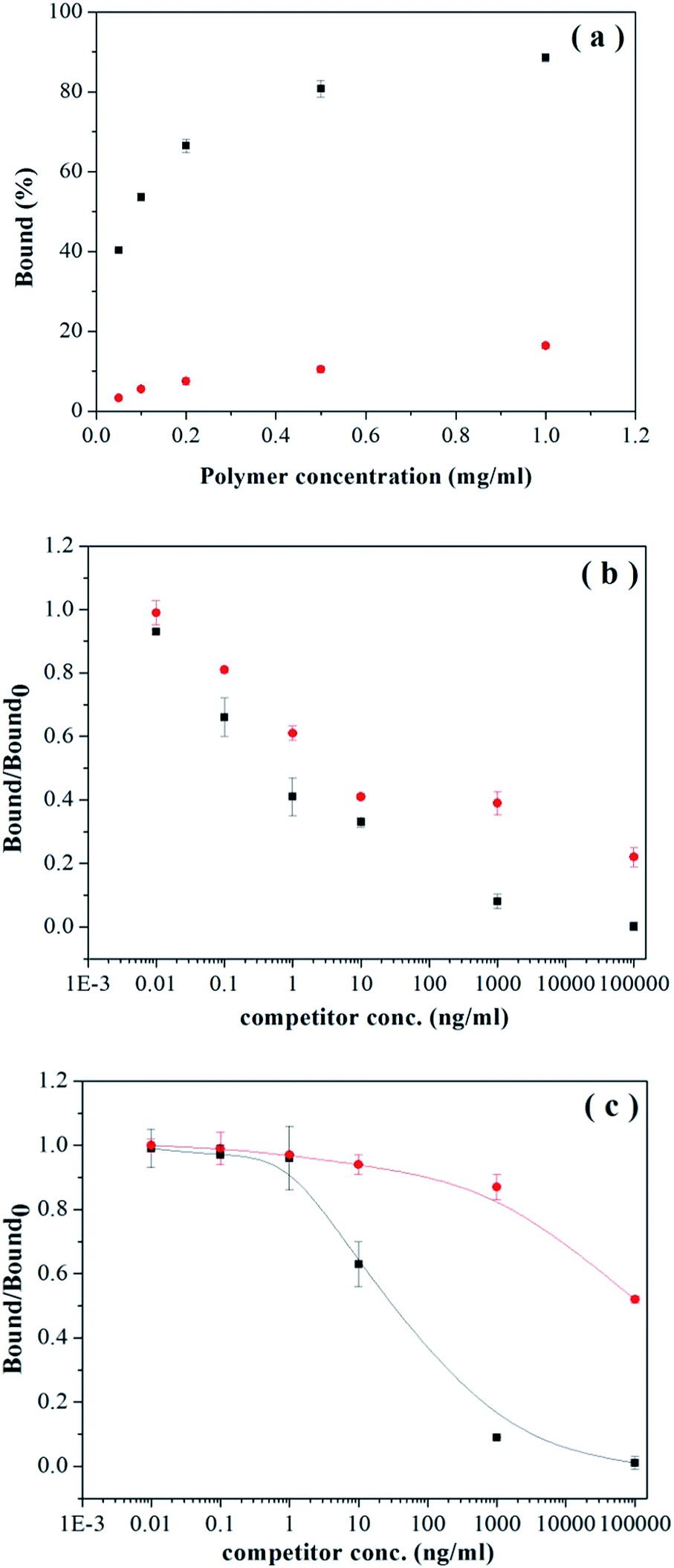 | ||
| Fig. 8 (a) Uptake of L-[N-methyl-3H]-nicotine (233 pM) by MIP-SH (■) and NIP-SH (●) in acetonitrile. (b) Displacement of L-[N-methyl-3H]-nicotine from 0.05 mg of MIP-SH by nicotine (■) and cotinine (●). (c) Displacement of L-[N-methyl-3H]-nicotine from 0.05 mg of MIP by nicotine (■) and cotinine (●). Bound and bound0 are the amount of the bound radioligand measured in the presence and absence of the competing compounds, respectively. | ||
The selectivity of MIP-SH for nicotine was further studied using competitive radioligand binding analysis.10 In this experiment, unlabelled nicotine and cotinine were used as competing molecules to displace tritium-labelled nicotine from the imprinted polymer particles. In principle, unlabelled nicotine should be the most potent inhibitor for the radioligand binding to the specific sites, as the test compound and the radioligand have the same structure. On the other hand, the structure of cotinine is different from nicotine, therefore cotinine should be less effective in displacing the tritium-labelled nicotine from the imprinted binding sites. Indeed, Fig. 8b shows that compared to nicotine, cotinine is obviously less effective in competing with the labelled nicotine to bind to MIP-SH particles.
Comparing the displacement curves in Fig. 8b with 8c leads to some striking observations. In Fig. 8c, the displacement curve of cotinine is more distant from nicotine than that found in Fig. 8b, implying that the MIP particles contain more high quality sites than the MIP-SH particles. This argument is also supported by the fact that for MIP-SH particles, radioligand displacement becomes obvious even in the low concentration range (0.01–1 ng mL−1). Since the RAFT agent contributes to specific binding, it should be possible to limit the aminolysis reaction on the surface of the imprinted particles (e.g. by using shorter reaction time, lower temperature, or smaller quantity of larger amine reagents), and at the same time to maintain high selectivity of the interior sites. Indeed, when the MIP particles were treated with the same aminolysis reaction for a shorter period (4 h), only 47% of the RAFT agent was converted into –SH groups (see ESI†). In competitive radioligand binding assays, these new MIP-SH particles displayed significantly higher selectivity for nicotine than cotinine (ESI, Fig. S1†). It is remarkable that the nicotine selectivity of these MIP-SH particles was not impaired by fluorescent labelling with NPM (ESI, Fig. S1†).
Conclusions
RAFT precipitation polymerization has been used successfully to synthesize uniform polymer microspheres with high selectivity for nicotine. Besides the well-known effect of the functional monomer MAA, we found that the RAFT agent CDB also contributed to the imprinting effect. For the first time, we demonstrated that new functionalities can be introduced to MIP microspheres by converting the RAFT agent through simple aminolysis reaction. The new thiol groups obtained provide a convenient tag to allow MIP microspheres to be conjugated with other molecules and functional materials. Many applications can be conceived for the new imprinted polymer microspheres, for example as basic building blocks to construct chemical sensors as well as to build polymer–enzyme conjugates for analytical applications. The use of thiol-functionalized MIP microspheres in surface enhanced Raman spectroscopy is being investigated in our laboratory and the results will be reported in a forthcoming paper.Acknowledgements
This work was supported by the Danish Council for Strategic Research (project FENAMI, DSF-10-93456) and the Swedish Research Council for Environment, Agricultural Sciences and Spatial Planning (FORMAS). We thank Dr Bruno Provstgaard Nielsen and Stig R. Knudsen from Fertin Pharma A/S for valuable scientific discussions.Notes and references
- G. Vlatakis, L. I. Andersson, R. Muller and K. Mosbach, Nature, 1993, 361, 645–647 CrossRef CAS PubMed.
- M. J. Whitcombe, C. Alexander and E. N. Vulfson, Trends Food Sci. Technol., 1997, 8, 140–145 CrossRef CAS.
- L. Ye and K. Mosbach, Chem. Mater., 2008, 20, 859–868 CrossRef CAS.
- S. A. Piletsky, N. W. Turner and P. Laitenberger, Med. Eng. Phys., 2006, 28, 971–977 CrossRef PubMed.
- E. Saridakis, S. Khurshid, L. Govada, Q. Phan, D. Hawkins, G. V. Crichlow, E. Lolis, S. M. Reddy and N. E. Chayen, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11081–11086 CrossRef CAS PubMed.
- B. Sellergren and C. J. Allender, Adv. Drug Delivery Rev., 2005, 57, 1733–1741 CrossRef CAS PubMed.
- G. Wulff, A. Sarhan and K. Zabrocki, Tetrahedron Lett., 1973, 4329–4332 CrossRef CAS.
- A. G. Mayes and K. Mosbach, Anal. Chem., 1996, 68, 3769–3774 CrossRef CAS PubMed.
- A. Poma, A. P. Turner and S. A. Piletsky, Trends Biotechnol., 2010, 28, 629–637 CrossRef CAS PubMed.
- K. Yoshimatsu, K. Reimhult, A. Krozer, K. Mosbach, K. Sode and L. Ye, Anal. Chim. Acta, 2007, 584, 112–121 CrossRef CAS PubMed.
- S. Boonpangrak, M. J. Whitcombe, V. Prachayasittikul, K. Mosbach and L. Ye, Biosens. Bioelectron., 2006, 22, 349–354 CrossRef CAS PubMed.
- G. Pan, Y. Zhang, X. Guo, C. Li and H. Zhang, Biosens. Bioelectron., 2010, 26, 976–982 CrossRef CAS PubMed.
- G. Pan, Y. Zhang, Y. Ma, C. X. Li and H. Q. Zhang, Angew. Chem., Int. Ed., 2011, 50, 11731–11734 CrossRef CAS PubMed.
- C. Gonzato, M. Courty, P. Pasetto and K. Haupt, Adv. Funct. Mater., 2011, 21, 3947–3953 CrossRef CAS.
- Y. K. Chong, T. P. T. Le, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1999, 32, 2071–2074 CrossRef CAS.
- A. Favier and M.-T. Charreyre, Macromol. Rapid Commun., 2006, 27, 653–692 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379–410 CrossRef CAS.
- S. Perrier and P. Takolpuckdee, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5347–5393 CrossRef CAS.
- G. Pan, B. Zu, X. Guo, Y. Zhang, C. Li and H. Zhang, Polymer, 2009, 50, 2819–2825 CrossRef CAS PubMed.
- H. Zhang, Eur. Polym. J., 2013, 49, 579–600 CrossRef CAS PubMed.
- Y. Ma, G. Pan, Y. Zhang, X. Guo and H. Zhang, Angew. Chem., Int. Ed., 2013, 52, 1511–1514 CrossRef CAS PubMed.
- C. Xu and L. Ye, Chem. Commun., 2011, 47, 6096–6098 RSC.
- C. Xu, X. Shen and L. Ye, J. Mater. Chem., 2012, 22, 7424–7433 Search PubMed.
- S. Hajizadeh, C. Xu, H. Kirsebom, L. Ye and B. Mattiasson, J. Chromatogr. A, 2013, 1274, 6–12 CrossRef CAS PubMed.
- X. Shen, C. Xu, K. M. A. Uddin, P.-O. Larsson and L. Ye, J. Mater. Chem. B, 2013, 1, 4612–4618 RSC.
- J. E. Henningfield and R. M. Keenan, J. Consult. Clin. Psychol., 1993, 61, 743–750 CrossRef CAS.
- J. Wu, K. S. Paudel, C. Strasinger, D. Hammell, A. L. Stinchcomb and B. J. Hinds, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 11698–11702 CrossRef CAS PubMed.
- K. S. Paudel, J. Wu, B. J. Hinds and A. L. Stinchcomb, J. Pharm. Sci., 2012, 101, 3823–3832 CrossRef CAS PubMed.
- L. Shahab, L. S. Brose and R. West, CNS Drugs, 2013, 27, 1007–1019 CrossRef CAS PubMed.
- R. Suedee, Int. J. Pharm. Sci. Rev. Res., 2013, 20, 235–268 CAS.
- M. I. Gibson, E. Frohlich and H. A. Klok, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4332–4345 CrossRef CAS.
- M. D. Rowe, B. A. G. Hammer and S. G. Boyes, Macromolecules, 2008, 41, 4147–4157 CrossRef CAS.
- B. Preinerstorfer, M. Lammerhofer and W. Lindner, J. Sep. Sci., 2009, 32, 1673–1685 CrossRef CAS PubMed.
- R. A. Sperling and W. J. Parak, Philos. Trans. R. Soc., A, 2010, 368, 1333–1383 CrossRef CAS PubMed.
- A. B. Lowe, Polym. Chem., 2010, 1, 17–36 RSC.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- R. I. Jølck, L. N. Feldborg, S. Andersen, S. M. Moghimi and T. L. Andresen, Adv. Biochem. Eng./Biotechnol., 2011, 125, 251–280 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra04741a |
| This journal is © The Royal Society of Chemistry 2014 |