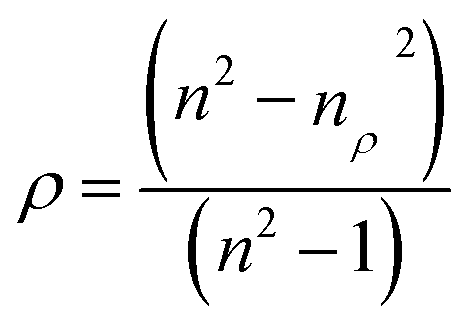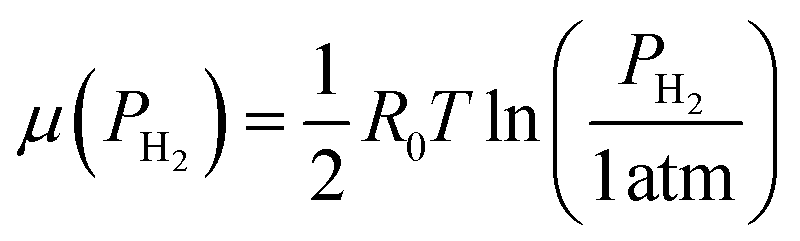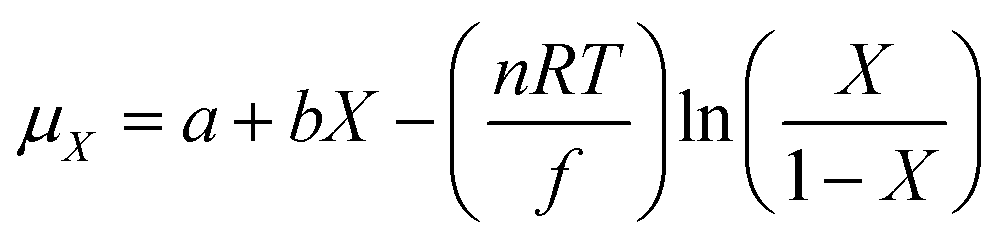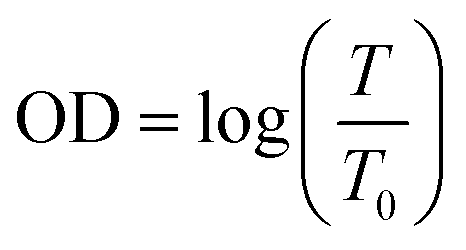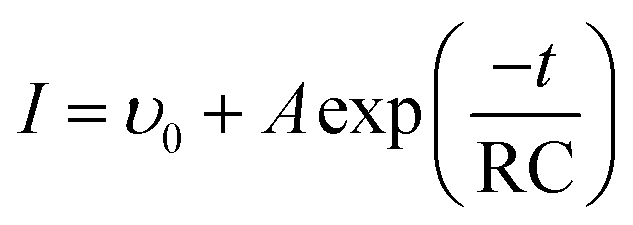DOI:
10.1039/C4RA03181D
(Paper)
RSC Adv., 2014,
4, 30300-30307
Engineering of coloration responses of porous WO3 gasochromic films by ultraviolet irradiation
Received
9th April 2014
, Accepted 2nd July 2014
First published on 2nd July 2014
Abstract
The coloration response of gasochromic films is crucial for gas sensors and solar energy cells. Based on a comparison of different post-treatments of WO3 gasochromic films, UV irradiation is found useful for fast coloration, in which fast exponential optical changes can be detected instead of a long activation delay process. Infrared spectroscopy, Raman spectroscopy, and X-ray photoelectron spectroscopy studies show that the gasochromic response of WO3 films depends on the ion and electron diffusion velocities, which can be engineered by altering the film porosities and conductivities. A new gasochromic model is proposed based on a resistor and capacitor (RC) circuit and the double-injection theory. According to this model, the slow coloration delay and fast exponential coloration should result from thermodynamic and electrochemical reactions, respectively. Our new model is also successfully used to build a link between two well-identified theoretical models.
1. Introduction
Gasochromic films that consist of a simple semiconductor layer (such as WO3) and a catalyst (such as Pd) are receiving considerable attention in optical hydrogen gas sensors,1–3 and large-scale solar cell applications.4 Several methods are used to prepare gasochromic films, such as sputtering,5 evaporation,6 sol–gel,7 electrochemical anodization8,9 and electrodeposition.10 Among these processes, the sol–gel technology is the most promising method for preparing large or irregular films because it is economical and requires minimal equipment. WO3 gasochromic films exhibit excellent selective sensitivity,11,12 deep coloration efficiency,7 and long-term stabilities.13,14 However, the slow coloration response of these films is still a limitation independent from the preparation method.5,7 An activation process is necessary to achieve rapid, stabilized gasochromic responses,7 which largely increases the uncertainty of gasochromic films in practical applications.
The gasochromism of amorphous tungsten oxide films is directly related to the double injection–extraction of ions and electrons,15–17 which can be expressed as:
| | |
xH+ + xe− + WO3 = HxWO3−x
| (1) |
This concept suggests that the coloring velocity rely on the ion (Ri) and electron (Re) conductivities.
The electrical conductivity of HxWO3 films has been identified to increase exponentially with increased x (when x > 0.2 at 300 K).18 On the other hand, ion diffusion relies on the porosity structure or ion diffusion channels.19,20 Hence, the optical response of gasochromic WO3 films can be improved by engineering their non-stoichiometry and porosity. Several studies based on the modifications of porous structure and non-stoichiometry have been conducted. Georg indicated that the coloration velocity largely depends on catalyst poisoning and water incorporation in porous WO3 films.21 Ranjbar proved the enhancement of the W5+/W6+ ratio by vanadium doping, which results in deeper and faster coloration.22 Modifying the porous structure of WO3, such as nanotextured Pt/WO3 thin films, can improve the sensitivity of its gas sensing response toward hydrogen.4,23
Under UV irradiation (UVI), proton and electrons enter amorphous WO3 films and form HxWO3 structures.24 Thus, inducing the UVI process can enable the adjustment of the conductivity of WO3 gasochromic films. The sol–gel method and thermal treatment are widely used to control the film porosities.25,26 These processes prompt us to use UVI and annealing approaches to identify an easy way of improving the gas-sensing performance of WO3 films.
In this study, the UVI method is used to improve the optical response of sol–gel WO3 films. Suitable irradiation time can remarkably decrease the activation process within 5 s. The structural changes caused by UVI are further studied by infrared (IR) spectra, elliptical polarized spectrograph, and X-ray photoelectron spectroscopy (XPS). Based on the experimental results, the effects of UVI on the improvement of gasochromic films are further discussed and explained.
2. Experimental
2.1. Preparation of gasochromic films
WO3 sols were prepared according to the method of Kudo.27 About 35 g of metallic W powder (99.8%) was added to 200 ml of H2O2 (30%) at room temperature in normal atmosphere. After the removal of impurities, the solution was evaporated (80 °C) until the sol color became transparent orange. PdCl2 was then added to the WO3 sol (0.2 mol L−1) as a catalyst in a concentration necessary to produce Pd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :W molar ratios of 1:50. All films were deposited onto glass slides or polished silicon slides by the dip-coating technique. UVI was carried out under a high-pressure Hg lamp (1 kW), producing two mercury lines at λ = 254.7 and 352.5 nm for different minutes. The samples were kept in air during irradiation, and the distance between the film and the light source was 5 cm.
:W molar ratios of 1:50. All films were deposited onto glass slides or polished silicon slides by the dip-coating technique. UVI was carried out under a high-pressure Hg lamp (1 kW), producing two mercury lines at λ = 254.7 and 352.5 nm for different minutes. The samples were kept in air during irradiation, and the distance between the film and the light source was 5 cm.
Two kinds of samples were prepared by the aforementioned method. The first sample was WO3 films without UVI; the other was irradiated for different minutes. All films (thickness = 250 nm) contained only two layers of WO3. The sample used for XPS was annealed at 50 °C for 1 h before UVI. Both glass slides for the samples were cleaned with acetone before XPS measurements.
2.2. Instruments and measurements
The transmission measurements of the films at the colored and bleached states were carried out on a UV-vis V-570 (Jasco Inc. series spectrometer) at 700 nm. A double-scan elliptical polarized spectrograph (ELLIP-A type, Shanghai FUFAN Positive Network Co., Inc.) was used to test the thickness and the refractive index. The spectrograph had a photo energy of 1.5–4.5 eV, wavelength resolution of 0.6 nm, and incidence control accuracy angle of 0.001° per pulse, where the accuracy of film thickness measurement can be controlled in 1 Å increments. The electron binding energy of WO3 was determined by XPS (PerkinElmer PHI5000c XPS/UPS) method.
3. Results and discussion
3.1. Optical response of the samples before and after UVI
The optical responses of WO3 films irradiated for 0, 10, 20, 30 min with UV lights are presented in Fig. 1(a). The film without UV treatment exhibits a long-term activation process that lasts for almost 240 s. This activation time can be largely reduced by UVI; however, UVI does no show a linear effect. The response of the sample illuminated for less than 10 min is still nearly 140 s, whereas that of the ones irradiated for more than 20 min can reach its saturated coloration within only 20 s without a slow activating step. A grating of the WO3 films was also designed to compare the UVI phenomena, as shown in the inset in Fig. 1(a), where the deep blue lines correspond to the parts irradiated by UV lights.
 |
| | Fig. 1 UV irradiation on the gasochromic responses of the WO3 films (a), IR spectra of the WO3 films with different irradiating times (b) and the ones annealed at 150 °C (c) and 450 °C (d) before and after the UV irradiation. | |
The IR spectra of the WO3 films irradiated for different times are shown in Fig. 1(b). WO3 films consist of three well-defined vibration groups: stretching modes of terminal (W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O at 975 cm−1), corner sharing (W–O–W at 642 cm−1),7,28 and edge-sharing (W–O–W at 802 cm−1) W–O modes.29 The bands at 642 cm−1 increase whereas those at 802 cm−1 and 975 cm−1 decrease as the irradiation process continues. These results indicate the formation of corner-sharing W–O–W bonds as well as the collapse of terminal WO and edge-sharing W–O–W bonds. Thus, the peroxo groups decompose and the structure of the edge-sharing WO6 groups, which prevail in sols and fresh xerogels, transform into a more condensed structure with corner-sharing WO6 units.7 These units are the same as the IR spectra of pristine sol–gel WO3 films and after completion of the first gasochromic coloring–bleaching cycle.30
O at 975 cm−1), corner sharing (W–O–W at 642 cm−1),7,28 and edge-sharing (W–O–W at 802 cm−1) W–O modes.29 The bands at 642 cm−1 increase whereas those at 802 cm−1 and 975 cm−1 decrease as the irradiation process continues. These results indicate the formation of corner-sharing W–O–W bonds as well as the collapse of terminal WO and edge-sharing W–O–W bonds. Thus, the peroxo groups decompose and the structure of the edge-sharing WO6 groups, which prevail in sols and fresh xerogels, transform into a more condensed structure with corner-sharing WO6 units.7 These units are the same as the IR spectra of pristine sol–gel WO3 films and after completion of the first gasochromic coloring–bleaching cycle.30
The IR spectral changes of the WO3 films annealed at 150 and 450 °C before and after UVI treatment were also examined, and the results are shown in Fig. 1(c) and (d). The films annealed at 150 °C exhibit similar structural changes, where the bands between 642 and 975 cm−1 both increase, and the band at 802 cm−1 disappears. This phenomenon has also been reported in the IR study of the coloring state of WO3 films in a previous study.30 Based on the analysis of Orel,7 the increased intensity of the bands between 642 and 802 cm−1 can both be assigned to the hydrogen-inserted WO3 films (W–OH). Therefore, the structural changes in Fig. 1(c) indicate the formation of corner-sharing W–O–W and W–OH bonds, as well as the collapse of edge-sharing W–O–W bonds. The orthorhombic phase of WO3 annealed at 450 °C can be clearly distinguished from the two strongest peaks at 728 and 807 cm−1, and no H2O characteristic can be detected. This result agrees with our XRD patterns, which show that the WO3 films exhibit an amorphous structure when annealed below 400 °C and a crystal structure when annealed at 450 °C.31 However, changes in the IR peaks caused by the UVI treatment are not obvious when the film is annealed at 450 °C.
The Raman spectra of WO3 before and after UVI are also examined (Fig. 2). The as-deposited WO3 films exhibit a single main peak at 802 cm−1 and a broad shoulder around 701 cm−1, assigned to a complex structure that consists of edge-sharing and corner-sharing W–O–W bonds.32–34 The structural transition of WO3 can be clearly distinguished after UVI from the two strongest peaks at 708 and 803 cm−1,35,36 where the edge-sharing W–O–W bonds largely decrease.
 |
| | Fig. 2 Raman spectra of WO3 films before and after the UV irradiation. | |
3.2. Surface morphology
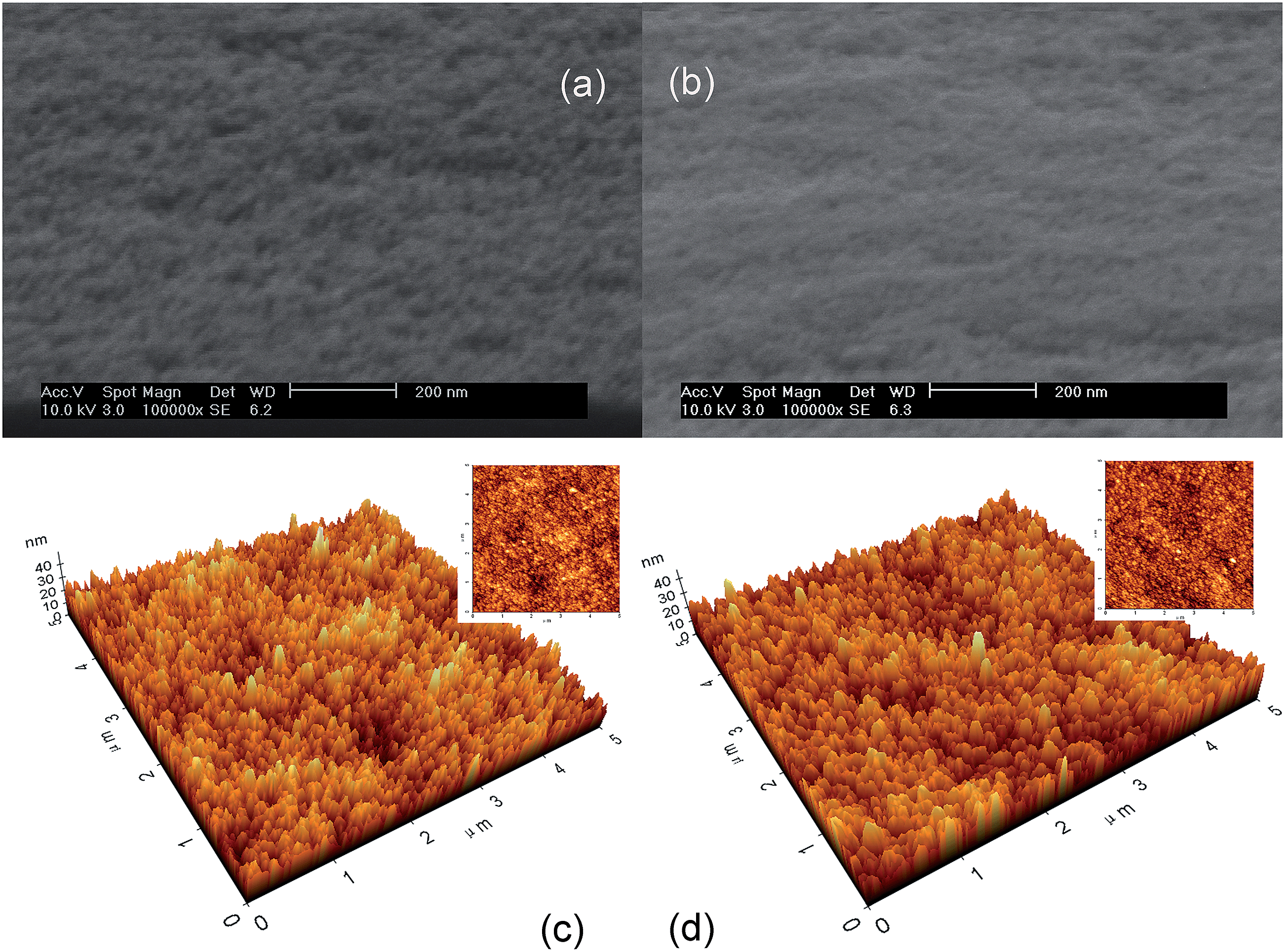
Scanning electron microscopy (SEM) and atomic force microscopy investigations were performed on both unirradiated and UV-irradiated WO3 films (Fig. 3). Typical images of the amorphous film surface with extensive homogeneity and without pore occlusions are observed. Our separated-roughness study shows that the clusters in the WO3 films are nearly spherical, with typical dimensions of 10–25 nm. Similar images are acquired for the UV-irradiated WO3 films, confirming that the macroporous structure is not affected by UVI. Hence, the microporous structures that cannot be easily detected by SEM spectra should be investigated.
 |
| | Fig. 3 SEM images of the WO3 films before (a) and after (b) the UV irradiation, AFM images of the WO3 films (c) and the UV illuminated WO3 films (d). | |
3.3. W 4f/O 1s XPS for UV-irradiated WO3 films
The electronic structure of the WO3 film surface is explored by XPS, as shown in Fig. 4 and Table 1. Fig. 4(a) shows the W4f spectra of the WO3 film before UVI. The band can be resolved into two bands at 37.85 and 35.75 eV for W 4f5/2 and W 4f7/2, respectively. The spin separation is 2.1 eV. This binding energy is assigned to the W(VI) O3 states.37 Four bands resolved from XPS spectra of the UV-treated films are observed, as shown in Fig. 4(b). The higher binding energies at 38.1 and 36.0 eV are attributed to the electrons of the WO3 without H+ insertion. The lower binding energy belongs to HxWO3.38 The x value is obtained by calculating the area ratio of the W5+ in W6+ states. The result indicates that the W5+ content is 14.6%; thus, x is equal to 0.17.
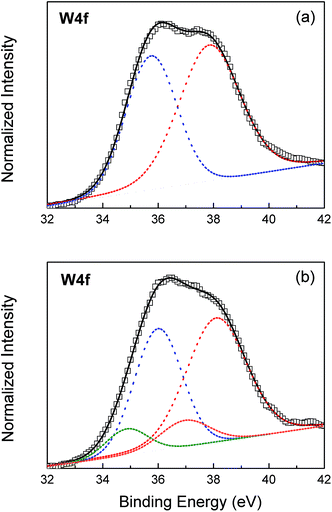 |
| | Fig. 4 W4f spectra and deconvolution curves of amorphous tungsten oxide films: as-deposited (a); irradiated by UV lights (b). | |
Table 1 Peak synthesis for W 4f-level XPS-spectrum of tungsten atoms
| |
Valence and spin separation |
Position (eV) |
ΔE (4f5/2 − 4f7/2) (eV) |
Width (eV) |
Area (%) |
| Before UV irradiation |
W6+ |
W 4f5/2 |
37.85 |
2.10 |
2.61 |
57.14 |
| W 4f7/2 |
35.75 |
2.17 |
42.86 |
| |
| After UV irradiation |
W6+ |
W 4f5/2 |
38.10 |
2.10 |
2.93 |
49.28 |
| W 4f7/2 |
36.00 |
1.87 |
36.14 |
| W5+ |
W 4f5/2 |
36.97 |
2.10 |
1.39 |
7.90 |
| W 4f7/2 |
34.87 |
1.46 |
6.69 |
3.4. Refractive index
The SEM results show that UVI is not effective against macroporous structures. Hence, microporous structures must be considered. The effect of UVI is further estimated by comparing the refractive indices of the annealed WO3 films before and after irradiation. Fig. 5 shows the refractive indices (n) of the investigated films as a function of the thermal treating temperature. The details are shown in Table 2. The values of n change over a wide range, i.e., from 1.90 (for the films annealed at 50 °C) to 2.17 (for those annealed at 450 °C). Characteristically, the refractive index of the WO3 films irradiated by UV is lower by 0.05 than that without UVI, whereas the gap of n between the WO3 samples annealed at other temperatures are close and become almost similar at 450 °C.
 |
| | Fig. 5 Comparison of refractive index and porosities of the WO3 annealed at different temperatures, before (a) and after (b) the UV irradiation. | |
Table 2 Refraction index and porosities of films annealed at various temperatures before and after the UV irradiation
| Temperature (°C) |
Refraction index |
Porosity |
| Before UVI |
After UVI |
ΔI(Ib−Ia) |
Before UVI (%) |
After UVI (%) |
ΔP(Pb−Pa) (%) |
| 50 |
1.930 |
1.907 |
0.023 |
27.38 |
29.73 |
2.35 |
| 150 |
2.058 |
2.026 |
0.032 |
13.77 |
17.26 |
3.48 |
| 250 |
2.100 |
2.090 |
0.010 |
9.12 |
10.24 |
1.12 |
| 350 |
2.136 |
2.128 |
0.008 |
5.06 |
5.97 |
0.91 |
| 450 |
2.180 |
2.175 |
0.005 |
0 |
0.58 |
0.58 |
The porosity of the nanofilms is significantly related to the refractive index, as shown by the following equation:39
| |
 | (2) |
where
nρ and
n are the refractive indices of the films and WO
3 crystal (2.18 is selected), respectively, and
ρ is the porosity of the films. Thus, the porosities of the films heated at different temperatures are obtained, as shown in
Fig. 5(b) and
Table 2. WO
3 films lost nearly 30% of their porosity during thermal treatment, resulting in a condensed structure. About 2.35% of the total porosity of the film annealed at 50 °C diminishes upon UV irradiation; this porosity lost further decreases with increased annealing temperatures. These results agree with the aforementioned IR spectra, where the photochromic effect induces the reaction of WO
3 with H
2O. Therefore, the UVI effect is weaker with decreased H
2O content.
4. Discussion
The structural changes resulting from UVI can be explained by the following photochromic mechanism:40,41| | |
WO3 + hν → WO*3 + h+ + e−
| (3) |
| | |
2H2O + 4h+ → O2 + 4H+
| (4) |
| | |
WO3 + xH+ + xe− → HxW(VI)1−xW(V)xO3
| (5) |
The reaction can be briefly described as follows. When WO3 films are irradiated by UV light, a special energy h creates holes and electrons pairs (eqn (3)). The created holes that exhibit high oxidizability react with the absorbed water on the surface or interior to produce protons (H+) (eqn (4)). The generated electrons are injected into the WO3 conduction band and then react with WO3 and H+ to form HxWxVW1−xVIO3 (eqn (5)), as shown in Scheme 1. Therefore, the changes in the W–O bonds from edge-sharing into corner-sharing, as well as the formation of W–OH are probably triggered by this photochromic process. The unchanged IR spectra of the film annealed at 450 °C can be explained by the absence of water resulting from the thermal treatment.
 |
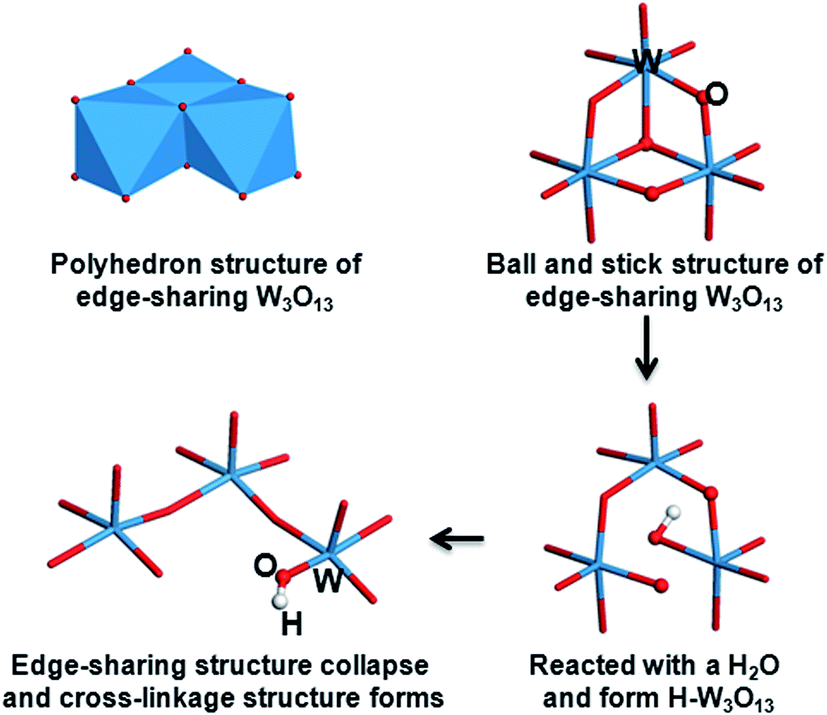
| | Scheme 1 The mechanism of photochromic coloration. | |
As aforementioned, the low valence of W in HxWO3 (x ≈ 0.17) is detected by XPS. The formation of corner-sharing W–O–W bonds and the collapsed edge-sharing of W3O13 clusters are also observed in the IR spectra. These structural transitions are illustrated in Scheme 2. A polyhedron structure of edge-sharing W3O13 that connects with one another by corner-sharing W–O–W bonds is formed in the as prepared films. The W3O13 cluster reacts with H2O molecule and forms a H–W3O13 unit when illuminated by UV light. Simultaneously, the edge-sharing W–O–W bonds break and transform into a cross-linked structure with only corner-sharing bonds remaining. The increase in porosity created by UVI may be due to the collapse of the W3O13 clusters during the reaction between the absorbed water and WO3 films. UVI also does not exert much effect on the WO3 film annealed at 450 °C. This finding agrees with the photochromic phenomena, indicating that hydrogen cannot be directly photoinjected into polycrystalline WO3 films because of the small specific surface area and weak absorptivity.24
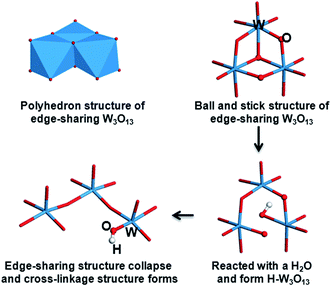 |
| | Scheme 2 Structure changes of WO3 clusters aroused from the UV irradiation. | |
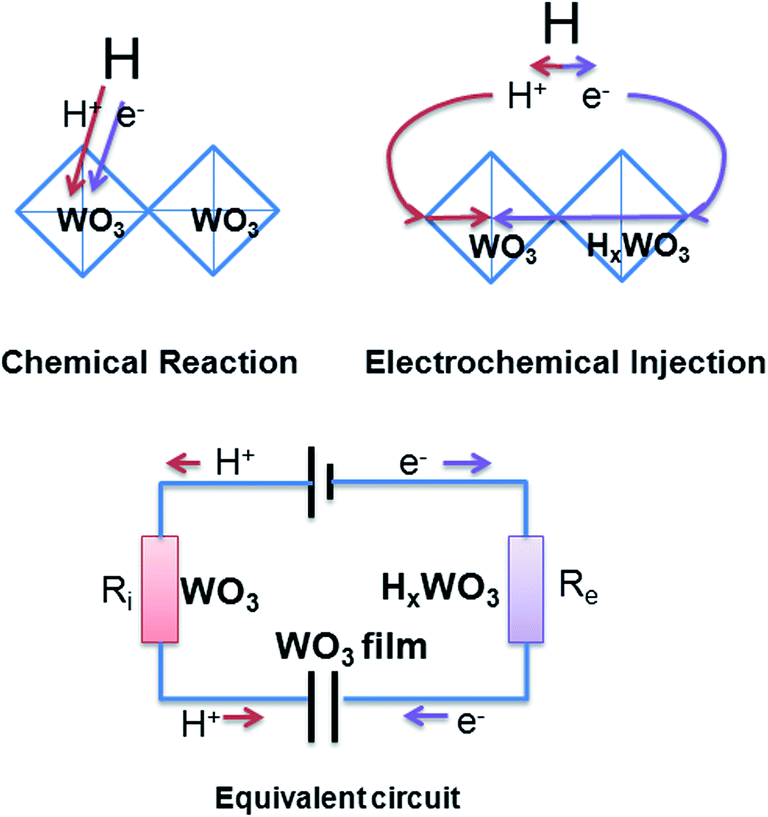
An electrochemical model is assumed based on electrical conductivity and ion diffusion to understand the complicated mechanism of gasochromic response (Scheme 3). According to the double-injection theory, the injection current depends on the ion (Ri) and electron (Re) resistant values in the series circuit, especially the larger ones. That means the double injection needs low ions and e− resistances. Before UVI, the WO3 film exhibits low Ri and high Re. At high Re values, the electrical conductivity is too small to form a return circuit. Thus, the insertion of hydrogen atom into HxWO3 can only be achieved by chemical reaction, which is mainly based on the Arrhenius equation (e−Ea/RT) and exhibits a slow reaction process. When films are irradiated by UV, the new formed HxWO3 gives low resistance for e−. So there is low Ri from WO3 and low Re from HxWO3 in the films and fast double injection is achieved. The insertion of hydrogen atom then performs in the electrochemical route and is exhibited as a RC circuit. The dissociated hydrogen atom is adsorbed onto the WO3 film surface and acts as the power source of the RC circuit. The WO3 films can serve as a Faraday capacitor. A hydrogen ion and an electron of the dissociated H atom are inserted into the WO3 lattice through Ri, which depends on the structural porosities from the anode, and through Re, which depends on the electrical conductivity from the cathode. Accordingly, the color center x exhibits the characteristic response of an RC circuit in exponential modes (e−t/RC), where R is the sum of Re and Ri.
 |
| | Scheme 3 Theoretical model. | |
According to this model, the electric potential of the HxWO3(x) capacitor is equal to the power voltage of H2 (μ(PH2)) at equilibrium.
According to the ideal gas law,5
| |
 | (7) |
where
R0 is the gas constant and
T is the temperature. Based on the theory and measurement of the chemical potential of amorphous H
xWO
3 by Crandall,
42 the chemical potential is expressed as:
| |
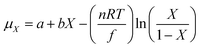 | (8) |
where
a,
b, and
n are constants, and
f is Faraday's constant. If the minimal interaction
42 (
bx) of the tungsten ions with their neighboring oxygen atoms is neglected, and
eqn (6) to (8) are combined, the color center
x of H
xWO
3 can be written as:
| |
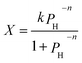 | (9) |
where
k and
n are constants and determined by the constants in
eqn (6) to (8).
Eqn (9) is the same as the experimental results of Chan.
15 Hence, the two different theoretical modes are linked based on our new assumption, which is also supported by a previous electrochemical study on gasochromism.
43
The complicated gasochromic response caused by the thermal and UVI treatments based on this model are next considered. The transmittances of the WO3 coloration are determined by color centers, which can be measured from optical density (OD) changes. The optical density is obtained from the following equation.
| |
 | (10) |
where
T and
T0 are the transmitted light transmittance at the colored and bleach states, respectively. The optical density decay is obtained and presented in
Fig. 6. The optical responses of each WO
3 films annealed at different temperatures exhibit an activation process, except for the films annealed at 50 °C and further irradiated by UV. Higher heating temperatures result in a more obvious activation process. UV treatment cannot overcome this activation process for the films annealed below 450 °C.
 |
| | Fig. 6 Single-exponential decay fitting of optical densities of WO3 films before (a) and after (b) the UV irradiation. | |
Based on our gasochromic response model, the OD decay for all samples before and after UVI is properly fitted by a single-exponential decay function as follows:
| |
 | (11) |
where
ϒ0 is the initial coloration velocity and
A is a constant determined by capacity
C and electric potential
E. The fitting results are shown in
Table 3. Two kinds of exponential process can be observed. One is a long-term exponential delay with a negative RC, and the other is a fast exponential coloration with a positive RC.
Table 3 Exponential decay fitting parameters of optical density of WO3 films before (a) and after (b) the UV irradiation
| Annealing temperature (°C) |
Before UV irradiation |
After UV irradiation |
| ϒ01 |
A1 |
RC1 |
R2 |
ϒ02 |
A2 |
RC2 |
R2 |
| 50 |
77.13 (42.84) |
−15.39 (27.77) |
−451.25 (208.01) |
0.997 (0.999) |
38.83 |
26.46 |
23.69 |
0.967 |
| 150 |
68.56 |
−6.67 |
−558.82 |
0.995 |
44.99 |
16.50 |
230.76 |
0.996 |
| 350 |
63.79 |
−2.22 |
−558.77 |
0.995 |
52.81 |
8.25 |
230.78 |
0.996 |
| 450 |
— |
— |
— |
|
62.90 |
−2.21 |
−558.60 |
0.995 |
Orel43 indicated that the proton conductivity of the WO3 film is always higher than the electronic conductivity. HxWO3 is also a semiconductor when x is less than the metal–insulator transition point (x0), and a metal when x is larger than x0.42 Hence, Re of the semiconductor before UVI is too large to build a return circuit, and a slow chemical reaction process can be observed. After UVI, the XPS results show that x in HxWO3 is 0.17, which is similar to the studies of Crandall.18 With this value, HxWO3 exhibits low resistance by which the series circuit in Scheme 3 is formed and a fast coloration response can be observed. The coloration process of deep coloration films without UVI can also be divided into an exponential delay in the beginning and exponential coloration in the subsequence process, such as the WO3 film annealed at 50 °C shown in Fig. 6(a). This result is attributed to the changes in electron density of state caused by the insertion of H. The WO3 films annealed at 450 °C show crystalline structures, which do not exhibit photochromism.44 Hence, the coloration activation process is still long even after UVI in Fig. 6(b).
The optical response significantly depends on the heating temperature regardless of UVI, as shown in Fig. 6 and Table 3, because ion diffusion relies on the structure porosity.19,20 For amorphous, the main interaction is at the defects and surfaces. For crystalline, the intercalation sites are mainly located inside of the bulks. Different intercalation sites arouse different diffusion barriers. Thermal treatment can largely influence the porosities and crystalline phase of WO3 films, resulting in large Ri value changes. Moreover, the HxWO3 can be decomposed into WO3−x and H2O.45 This RC model we proposed here mainly focused on the fast H intercalation process. Slow decomposition and oxygen diffusion are assigned to the high R situation, where the activation energy and the diffusion constant should be further studied.
5. Conclusions
UVI can largely improve the gasochromic response of WO3 films prepared by sol–gel methods. Under irradiation, the porosities of WO3 films largely increase, and the formation of HxWO3 (where x = 0.17) can be detected. The optical response of WO3 gasochromic films exhibits two coloration responses: exponential delay and exponential coloration. Further investigation on the thermal treatments reveals that the porosity can only increase the coloration velocity but cannot overcome the activation in exponential delay. A new gasochromic model based on RC circuits is proposed according to the double-injection concepts, which very well agrees with the single-exponential decay fitting of the optical density. This model is also successfully used to build a link between the chemical potential μx and hydrogen insertion coefficient x in two separate theories.
Acknowledgements
The authors gratefully acknowledge the financial support by National Natural Science Foundation of China (grant numbers 51272179, 51102183), Shanghai Committee of Science and Technology (11nm0501300, 13JC1408700), National high-tech R-D program of China (863 program) (grant no. 2013AA031801), Bayer Science & Education Foundation.
References
- L. F. Zhu, J. C. She, J. Y. Luo, S. Z. Deng, J. Chen and N. S. Xu, J. Phys. Chem. C, 2010, 114, 15504–15509 CAS.
- J. Z. Ou, M. H. Yaacob, J. L. Campbell, M. Breedon, K. Kalantar-zadeh and W. Wlodarski, Sens. Actuators, B, 2012, 166, 1–6 CrossRef PubMed.
- M. H. Yaacob, M. Z. Ahmad, A. Sadek, J. Z. Ou, J. Campbell, K. Kalantar-zadeh and W. Wlodarski, Sens. Actuators, B, 2013, 177, 981–988 CrossRef CAS PubMed.
- M. Ranjbar, N. T. Garavand, S. M. Mahdavi and A. I. Zad, Sol. Energy Mater. Sol. Cells, 2010, 94, 201–206 CrossRef CAS PubMed.
- A. Georg, W. Graf, R. Neumann and V. Wittwer, Solid State Ionics, 2000, 127, 319–328 CrossRef CAS.
- D. Gogova, L. K. Thomas and B. Camin, Thin Solid Films, 2009, 517, 3326–3331 CrossRef CAS PubMed.
- B. Orel, U. O. Krasovec, N. Groselj, M. Kosec, G. Drazic and R. Reisfeld, J. Sol-Gel Sci. Technol., 1999, 14, 291–308 CrossRef CAS.
- J. Z. Ou, S. Balendhran, M. R. Field, D. G. McCulloch, A. S. Zoolfakar, R. A. Rani, S. Zhuiykov, A. P. O'Mullane and K. Kalantar-zadeh, Nanoscale, 2012, 4, 5980–5988 RSC.
- J. Z. Ou, R. A. Rani, S. Balendhran, A. S. Zoolfakar, M. R. Field, S. Zhuiykov, A. P. O'Mullane and K. Kalantar-zadeh, Electrochem. Commun., 2013, 27, 128–132 CrossRef CAS PubMed.
- W. C. Hsu, C. C. Chan, C. H. Peng and C. C. Chang, Thin Solid Films, 2007, 516, 407–411 CrossRef CAS PubMed.
- K. Galatsis, Y. X. Li, W. Wlodarski and K. Kalantar-zadeh, Sens. Actuators, B, 2001, 77, 478–483 CrossRef CAS.
- L. G. Teoh, I. M. Hung, J. Shieh, W. H. Lai and M. H. Hon, Electrochem. Solid-State Lett., 2003, 6, G108–G111 CrossRef CAS PubMed.
- D. Z. Li, G. M. Wu, G. H. Gao, J. Shen and F. Q. Huang, ACS Appl. Mater. Interfaces, 2011, 3, 4573–4579 CAS.
- G. H. Gao, J. D. Wu, G. M. Wu, Z. H. Zhang, W. Feng, J. Shen and B. Zhou, Sens. Actuators, B, 2012, 171, 1288–1291 CrossRef PubMed.
- C. C. Chan, W. C. Hsu, C. C. Chang and C. S. Hsu, Sens. Actuators, B, 2011, 157, 504–509 CrossRef CAS PubMed.
- S. H. Lee, H. M. Cheong, P. Liu, D. Smith, C. E. Tracy, A. Mascanrenhas, J. R. Pitts and S. K. Deb, J. Appl. Phys., 2000, 88, 3076–3078 CrossRef CAS PubMed.
- S. Yamamoto, A. Inouye and M. Yoshikawa, Nucl. Instrum. Methods Phys. Res., Sect. B, 2008, 266, 802–806 CrossRef CAS PubMed.
- R. S. Crandall and B. W. Faughnan, Phys. Rev. Lett., 1977, 39, 232–235 CrossRef CAS.
- S. H. Lee, R. Deshpande, P. A. Parilla, K. M. Jones, B. To, A. H. Mahan and A. C. Dillon, Adv. Mater., 2006, 18, 763–766 CrossRef CAS.
- A. Vértes and R. Schiller, J. Appl. Phys., 1982, 54, 199–203 CrossRef PubMed.
- A. Georg, W. Graf, R. Neumann and V. Wittwer, Sol. Energy Mater. Sol. Cells, 2000, 63, 165–176 CrossRef CAS.
- M. Ranjbar, S. M. Mahdavi and A. I. Zad, Sol. Energy Mater. Sol. Cells, 2008, 92, 878–883 CrossRef CAS PubMed.
- M. H. Yaacob, M. Breedon, K. Kalantar-Zadeh and W. Wlodarski, Sens. Actuators, B, 2009, 137, 115–120 CrossRef PubMed.
- T. He and J. N. Yao, Prog. Mater. Sci., 2006, 51, 810–879 CrossRef CAS PubMed.
- U. Opara-Krasovec, R. Jese, B. Orel, J. Grdadolnik and G. Drazic, Monatshefte Fur Chemie, 2002, 133, 1115–1133 CrossRef CAS.
- J. Gallardo, P. Galliano and A. Duran, J. Sol-Gel Sci. Technol., 2000, 19, 393–397 CrossRef CAS.
- T. Kudo, H. Okamoto, K. Matsumoto and Y. Sasaki, Inorg. Chim. Acta, 1986, 111, L27–L28 CrossRef CAS.
- T. Nanba, S. Takano, I. Yasui and T. Kudo, J. Solid State Chem., 1991, 90, 47–53 CrossRef CAS.
- B. Orel, N. Groselj, U. O. Krasovec, R. Jese and A. Georg, J. Sol-Gel Sci. Technol., 2002, 24, 5–22 CrossRef CAS.
- U. O. Krasovec, B. Orel, A. Georg and V. Wittwer, Sol. Energy, 2000, 68, 541–551 CrossRef CAS.
- J. C. Shi, G. M. Wu, S. W. Chen, J. Shen, B. Zhou and X. Y. Ni, Chem. J. Chin. Univ., 2007, 28, 1356–1360 CAS.
- T. Nanba, Y. Nishiyama and I. Yasui, J. Mater. Res., 1991, 6, 1324–1333 CrossRef CAS.
- P. Delichere, P. Falaras, M. Froment, A. Hugotlegoff and B. Agius, Thin Solid Films, 1988, 161, 35–46 CrossRef CAS.
- C. Guery, C. Choquet, F. Dujeancourt, J. M. Tarascon and J. C. Lassegues, J. Solid State Electrochem., 1997, 1, 199–207 CrossRef CAS.
- J. Y. Luo, S. Z. Deng, Y. T. Tao, F. L. Zhao, L. F. Zhu, L. Gong, J. Chen and N. S. Xu, J. Phys. Chem. C, 2009, 113, 15877–15881 CAS.
- Y. P. He and Y. P. Zhao, J. Phys. Chem. C, 2008, 112, 61–68 CAS.
- Y. F. Lu and H. Qiu, J. Appl. Phys., 2000, 88, 1082–1087 CrossRef CAS PubMed.
- J. I. Jeong, J. H. Hong, J. H. Moon, J. S. Kang and Y. Fukuda, J. Appl. Phys., 1996, 79, 9343–9348 CrossRef CAS PubMed.
- G. M. Wu, J. Wang, J. Shen, T. H. Yang, Q. Y. Zhang, B. Zhou, Z. H. Deng, B. Fan, D. P. Zhou and F. H. Zhang, Mater. Res. Bull., 2001, 36, 2127–2139 CrossRef CAS.
- T. He and J. N. Yao, J. Photochem. Photobiol., C, 2003, 4, 125–143 CrossRef CAS.
- Y. Zhang, S. H. Lee, A. Mascarenhas and S. K. Deb, Appl. Phys. Lett., 2008, 93, 203508 CrossRef PubMed.
- R. S. Crandall, P. J. Wojtowicza and B. W. Faughnana, Solid State Commun., 1976, 18, 1409–1411 CrossRef CAS.
- B. Orel, N. Groselj, U. O. Krasovec, M. Gabrscek, P. Bukovec and R. Reisfeld, Sens. Actuators, B, 1998, 50, 234–245 CrossRef CAS.
- E. Kikuchi, K. Iida and A. Fujishima, J. Electroanal. Chem., 1993, 351, 105–114 CrossRef CAS.
- J. Z. Ou, M. H. Yaacob, M. Breedon, H. D. Zheng, J. L. Campbell, K. Latham, J. du Plessis, W. Wlodarski and K. Kalantar-Zadeh, Phys. Chem. Chem. Phys., 2011, 13, 7330–7339 RSC.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.