
Unusual post-translational protein modifications: the benefits of sophistication
Boddepalli Ravikiran
and
Radhakrishnan Mahalakshmi
 *
*
Molecular Biophysics Laboratory, Department of Biological Sciences, Indian Institute of Science Education and Research, Bhopal, India. E-mail: maha@iiserb.ac.in
First published on 15th July 2014
Abstract
The proteome of an organism represents the work force that is responsible for cellular activities, regulation and survival. Subsequent to synthesis and folding, there is growing evidence that proteins can undergo several novel and previously unknown post-translational modifications (PTMs) that are structurally and functionally significant. Non-disulphide backbone–side chain or side chain–side chain covalent bonds and supplementary modifications that chiefly generate catalytic centres and render proteinaceous enzymes functionally autonomous, are highlighted in this review. Currently known biosynthetic mechanisms derived using modern methodology for the identification of such PTMs are discussed.
 Boddepalli Ravikiran | Boddepalli Ravikiran was born in Srikakulam, India, in 1992. He attended Little Angels high school and Visakha Junior College in Visakhapatnam, India before moving to Bhopal, India to pursue his BS-MS (dual) degree program at IISER Bhopal in 2009. He carried out his final year thesis work under the guidance of Dr R. Mahalakshmi, IISER Bhopal from 2013 to 2014. He received his BS-MS (dual) degree in Biological Sciences from IISER Bhopal in 2014. He was awarded the INSPIRE fellowship during his BS-MS program. He is planning to pursue a career in financial management and is currently preparing for various national level examinations to pursue a postgraduate degree in financial management. |
 Radhakrishnan Mahalakshmi | Radhakrishnan Mahalakshmi received her M.S. and Ph.D. from the Indian Institute of Science, Bangalore, in 2003 and 2006, and then carried out her postdoctoral research at the (now) Sanford–Burnham Medical Research Institute and La Jolla Institute for Allergy and Immunology, San Diego, U.S.A. She joined the Indian Institute of Science Education and Research at Bhopal as Assistant Professor in 2009. Her research interests include: (i) mechanistic understanding of the inter-relationship between structure–function–regulation of transmembrane proteins and their implications in apoptosis and neurodegeneration. (ii) Deducing membrane protein folding pathways and understanding the thermodynamic and kinetic components governing these processes. (iii) Development of peptide mimetics to study protein stability and enzymatic catalysis. She is the recipient of the Ramalingaswami Fellowship, Innovative Young Biotechnologist Award, INSA medal for Young Scientists and the Wellcome Trust/DBT India Alliance Intermediate Fellowship. |
Introduction
The work force responsible for the cellular activities, regulation and survival of any organism is its proteome. Unlike nucleic acids, the innumerable permutations possible with the proteinogenic amino acids confer on proteins a variety of assorted and discrete functions that determine the diversity among living organisms. Once synthesized, folding of the protein to its optimal two- and three-dimensional structure (with varied levels of order or disorder), and possibly oligomerization, is required for its downstream activity, which may be enzymatic, structural or regulatory. During or subsequent to the process of synthesis, a protein molecule undergoes a variety of post-translational modifications (PTMs) with varied levels of complexity, which are vital for structure and downstream enzymatic, structural or regulatory activity. Disulphide bond formation is the most common and widely prevalent of all known PTMs.The six major classes of enzymes can easily participate in five broad classes of reactions, of which oxidation–reduction reactions and group transfer processes largely require the presence of small molecules called cofactors that act as the enzyme's chemical teeth. Cofactors (or coenzymes, if they are organic molecules) vary in their physicochemical properties, ranging from metal ions and organic molecules to prosthetic groups such as porphyrins and vitamins. Standard textbooks define cofactors (or coenzymes) as small molecules that bind to protein active sites and aid in catalysis. Cofactors can be separated from the protein molecule to give rise to the inactive apoenzyme form of the protein. The presence of cofactors for active functioning of the enzyme was for a long time considered so mandatory that when histidine decarboxylase from Lactobacillus sp. was found, in the early 1990s, to function in the absence of PLP (pyridoxal phosphate, the normal cofactor used), it came as a surprise for the scientific community. The crystal structure of this unusual enzyme, published in 1993, for the first time revealed an unusual modification that efficiently functions as an analogue of the normal cofactor, thereby completely dispensing with the need for PLP.1 The cofactor, generated by autocatalytic serinolysis of Ser 82 in the proenzyme, forms a Schiff base with the substrate and facilitates histidine decarboxylation.
Over the last decade, a large number of unusual PTMs in proteins have been unearthed, which include newly identified cross-links between amino acid side chains, thanks to high-resolution structures, combined with biochemical studies. With the advent of modern techniques and methodology for the study of proteins at the molecular level, including mass spectrometry, high-resolution X-ray diffraction and nuclear magnetic resonance (NMR) spectroscopic methods, it has now become possible to discover and characterize several novel and previously unknown protein PTMs, and map functional and structural significance to these modifications. The only well-known side chain cross-link in proteins is the disulphide. Identification and characterization of novel, non-disulphide covalent bonds, which include carbon–carbon, carbon–sulphur, nitrogen–sulphur, nitrogen–carbon and oxygen–carbon bonds between amino acid side chains at protein active sites, has paved the way for newer and more stable post-translational modifications in proteins. Such PTMs not only contribute to additional diversity in these biomolecules but also play a key role in many cellular processes catalysed by enzymes.
While changes such as acetylation, glycosylation, lipidation, phosphorylation, etc., are well known and commonly observed in several proteins across organisms, this new class of covalent post-translational modifications (PTMs) is increasingly being recognized in a small class of proteins. This unique class of PTMs appositely adopted by nature includes non-disulphide peptide backbone–side chain or side chain–side chain covalent bonds that chiefly generate catalytic centres and render such proteinaceous enzymes functionally autonomous, and thereby independent of external organic cofactors. These potent intrinsic cofactors are primarily derived from and composed of amino acids like tryptophan, tyrosine, cysteine, histidine and lysine.
This review attempts to summarize such unique PTMs, probe their possible function and discuss the implications of such modifications for the design of artificial enzymes. Proteins bearing such unusual covalent modifications are largely enzymes, but also include a smaller, yet significant, class of structural proteins, all of which we categorize into the following sections:
1. Proteins that contain a side chain–side chain cross-link and are a broader class of enzymes, including oxidases, peroxidases, catalases, etc. These cross-links are observed between the side chains of amino acids present near the active site, and are usually coordinated with a metal ion. These cross-links either provide structural stability to the active site and/or participate in the catalytic reaction itself. Some examples we discuss include cytochrome c oxidase, catechol oxidase, catalase peroxidase and galactose oxidase.
2. Enzymes that contain side chain–side chain or side chain–backbone cross-links or other backbone alterations with supplementary modifications including dehydrogenases, oxidases, etc. The cofactors are amino acid-derived quinones that are present at the active site and provide structural stability to the enzymes' active site and participate in the catalytic activity. Some examples we discuss include lysyl oxidase, methanol dehydrogenase and a few amine dehydrogenases.
3. A small class of non-enzymatic proteins, which contain unusual covalent non-disulphide cross-links, providing special characteristics and inter-subunit structural stability to the molecule. Examples of this class include ranasmurfin, green fluorescent protein (GFP) and collagen. Such modifications are not just confined to larger proteins and enzymes, but are also detected in small, ribosomally encoded peptides, examples of which include a few Amanita toxins and lantibiotics.
In this framework, we review the biochemical pathways by which such unusual post-translational modifications are synthesized, the structures and local conformational geometry of such modifications, the functional implications and the recent advancements in the field, which are increasingly fascinating to examine. While we have tried to include exciting findings and known examples of unusual PTMs, there may still be a few modifications that have escaped our notice, or have already been covered extensively elsewhere,2 and are therefore not dealt with in detail here.
Side chain cross-links in enzymes
Enzymes catalyse nearly all known biochemical pathways within any living organism, and are vital for the sustenance of life forms. Many enzymes are assisted by cofactors for their functioning, which may range from inorganic metal ions, organic molecules (vitamins, nucleosides, etc.) to molecules that are nearly 1 kDa in molecular weight. It is known that some enzymes require multiple cofactors; for instance, pyruvate dehydrogenase utilizes five cofactors and a metal ion.3 Addition of such extrinsic components to an enzyme renders it functional. Recently, however, with the advent of advanced biochemical and biophysical methods, a new class of PTMs which result in amino acid-derived cofactors were discovered.2c Such cofactors are of great biological significance, as these structures help expand the range of chemical reactions involving amino acids, as well as creating new structural motifs that provide structural stability to the active site.Oxidases
GO is a copper metalloenzyme which contains an unusual tyrosine–cysteine thio–ether cross-link in its active site. This cross-link serves as an intrinsic cofactor which helps in transferring the dihydrogen, unlike other enzymes which employ extrinsic cofactors like flavins, nicotinamides and quinones. The active site is a copper complex in which the copper is surrounded by Tyr 272, Tyr 495, His 496 and His 581. Tyr 272 Cε is cross-linked to the side chain sulphur of Cys 228 (residue numbering is that of GO from Fusarium sp.) (Fig. 1).4
 | ||
| Fig. 1 Ribbon diagram of galactose oxidase, highlighting the Cys–Tyr cross-link.4 | ||
The biogenesis of the cofactor is only dependent on the presence of Cu1+ and molecular oxygen.4a Kinetics data comparing the cofactor synthesis in the presence of Cu1+ and Cu2+ supports the sole requirement for monovalent copper (since the divalent form is generated during the reaction, as described below). In this process, Cu1+ initially binds to a pre-organized active site and coordinates with tyrosine, forming a reduced metal complex. O2 reacts directly with the reduced metal complex to form a Cu2+–superoxide, which is a reactive adduct. This oxygenated adduct abstracts a hydrogen atom from the active site Cys 228, resulting in the formation of a thiyl radical, which adds to the Tyr 272 aromatic ring. The addition of this thiyl radical to tyrosine gives rise to a non-conjugated system in the phenolic side chain. The Tyr aromaticity is then restored by deprotonation and reduction of Cu2+ to Cu1+.2f Hence, during the reaction turnover, this Cu1+ form at the active site corresponds to the reduced form of the protein. This reacts further with O2. The oxidised Cu–tyrosyl radical complex is subsequently formed by the action of O2.2f The reaction is presented in Fig. 2.
 | ||
| Fig. 2 Proposed mechanism of galactose oxidase Tyr–Cys cofactor. Figure re-drawn with permission from Whittaker.2f | ||
The Tyr–Cys thio–ether bond influences the stability, reduction potential and catalytic efficiency of the GO active site. It has been reported that the lack of this linkage has only a minimal effect on the Cu coordination at the active site. However, it influences the π-stacking interaction between the Cu-bound Tyr 272, and the nearby Trp 290, which is important to keep the active site geometry intact. This is evident by comparison of the active site geometries of the precursor and mature proteins, available from crystal structures of GO from Fusarium sp.4
Furthermore, the cross-link acts as an independent redox cofactor, allowing easy electron delocalisation. The Cu(II)/Cys–Tyr* cofactor carries out a 2e− oxidation of primary alcohols to aldehydes via a radical mechanism.4a The cross-link also reduces the reduction potential by about 75 mV, which is sufficient to modulate the reactivity so that the activated C–H bond of the alcohol is attacked preferentially by the tyrosine.2h The mechanism by which the molecular oxygen is reduced to hydrogen peroxide is shown in Fig. 3.2e,f The protein shows activity only in the mature cross-linked form.
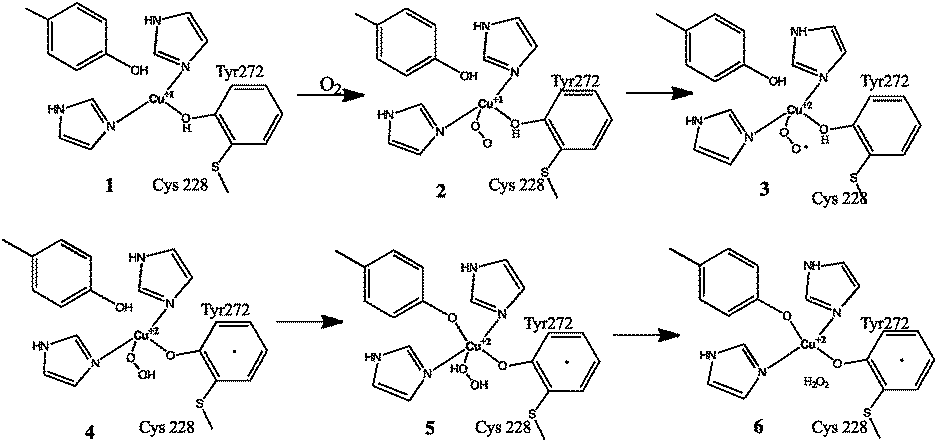 | ||
| Fig. 3 Dioxygen reduction mechanism of galactose oxidase. Figure re-drawn with permission from Whittaker.2f | ||
 | ||
| Fig. 4 Ribbon diagram of cytochrome c oxidase (right), which contains the His–Tyr cross-link at the oxygen binding site. Shown as stick representations (left) are the oxidised and reduced forms of the active site, highlighting the importance of the cross-link in binding molecular oxygen.5 | ||
One of the reactions catalysed at the active centre of cytochrome c oxidase is dioxygen activation. It is both important and yet equally difficult to transfer electrons and protons successively to dioxygen (O2), while ensuring no formation or release of the superoxide anion radical ˙O2−, peroxide O22− or the hydroxyl radical ˙OH during the peroxide cleavage, since all these molecules are deleterious to the cell. The His–Tyr linker plays a very important role in achieving this. Further, it is believed that the pK of the hydroxyl group of Tyr is significantly lower than in free Tyr, due to the cross-link, which is important for Tyr to act as a proton donor in the reduction of molecular oxygen by the enzyme.7 The mechanism which has been proposed for the peroxide cleavage is described in detail elsewhere.8
Using chemical model studies, it has also been shown that the His–Tyr link has a role in the proton pumping action of the enzyme and plays a role in gating the K and D channels.9 The K and D channels present in the protein facilitate the intra-protein proton transfer by connecting the active site where the dioxygen is reduced to the internal aqueous phase. The His–Tyr link therefore acts as an electron/proton acceptor/donor and helps in the K and D channel gating.
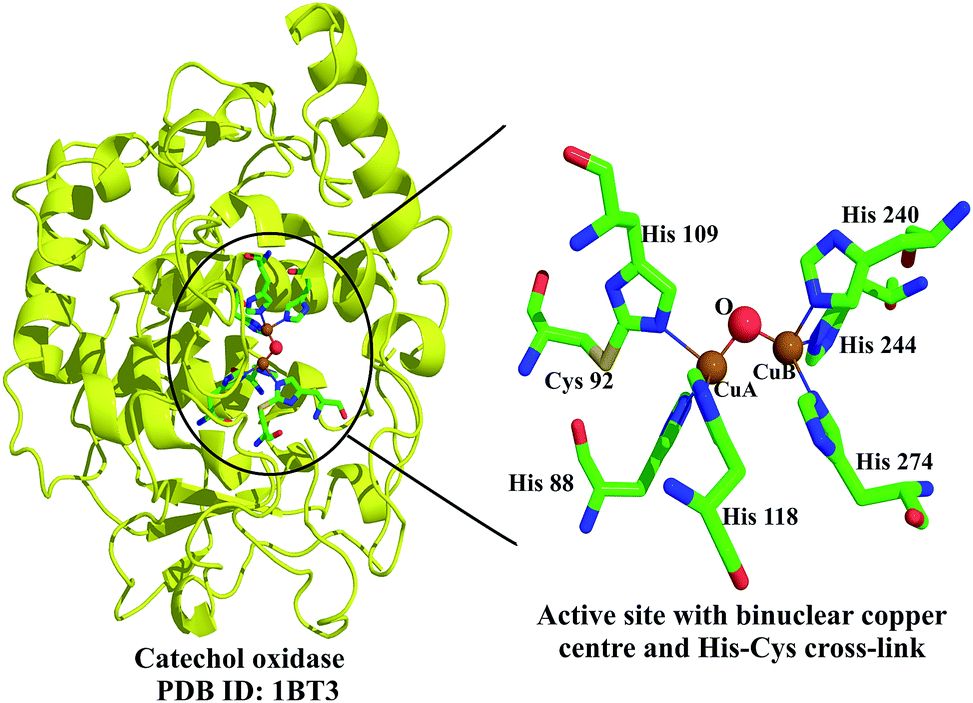 | ||
| Fig. 5 The unusual Cys–His cross-link in sweet potato catechol oxidase (shown on the left in a ribbon diagram) is highlighted on the right.10 The active site contains two copper atoms (CuA and CuB) that are coordinated by three histidines each. | ||
Peroxidases
 | ||
| Fig. 6 Ribbon diagram of catalase peroxidase highlighting the location of the cross-link and the active site, shown in stick representation.13 | ||
 | ||
| Fig. 7 Proposed role of the MYW adduct in the catalytic activity of catalase peroxidase. Figure adapted with permission from Zhao et al.2g Copyright (2010) American Chemical Society. | ||
A study that attempted to generate mutational variants of CcP, in attempts to engineer novel enzymatic activity into CcP, replaced the distal His 52 with Tyr.15 The protein was crystallized in the P212121 space group and solved to a 1.65 Å resolution.15 Surprisingly, the bond distance obtained between Tyr 52 Cε1 and the neighbouring Trp 51 Nε1 was found to be <1.6 Å, which, after structure refinement, was seen to give a bond distance of 1.48 Å, clearly indicating a covalent link between Tyr and Trp side chains. Since CcP was crystallized along with the redox-active Fe3+–porphyrin, a crystal structure solved using the redox-inactive Zn2+–porphyrin not only lacked the cross-link but also positioned Tyr 52–60° away from the active site.15
The unique Tyr–Trp cross-link was therefore established as a crystallization artefact, formed as a result of an old batch of 2-methyl-2,4-pentanediol (MPD) used in the crystallization set-up (MPD generates breakdown products such as peroxides). Using an elegant combination of conditions involving zinc or iron, and treatment with H2O2, Bhaskar et al. demonstrated that both iron and peroxide are important for the cross-linking process.15 Since aromatic radicals are inert to nucleophilic attack, formation of the cross-link requires concurrent oxidation at both sites, and is likely to occur via a peroxidic intermediate.
Interestingly, the cross-link results in a highly pyramidal intermediate, with the Trp Nε1–Tyr Cε1 bond bent ∼64° from the aromatic plane (Fig. 8). A similar observation has been made earlier only in the case of the rebeccamycin class of indolocarbazole glycosides, where the indolic N–sugar bond is ∼27° out of the aromatic plane.16 Semi-empirical calculations carried out suggested a small energy cost of ∼2 kcal mol−1 for a 30° deformation, but an immense 9.8 kcal mol−1 for the observed 60° deformation in CcP.15 While the implications of this modification in vivo are still unclear, CcP may serve as an excellent model system for studies on unusual cross-links in proteins, and their associated stabilization in a three-dimensional protein scaffold. Furthermore, this experimental artefact serves as the only known example of a Tyr–Trp cross-link in proteins.
 | ||
| Fig. 8 Superposition of the crystal structures of cytochrome c peroxidase highlighting the residues undergoing cross-linking. In the absence of a strong oxidising environment, the cross-link is not observed (blue). In the presence of iron and peroxide, the cross-link is formed. The Trp Nε1–Tyr Cε1 bond is bent ∼64° from the aromatic plane.15 | ||
Catalases
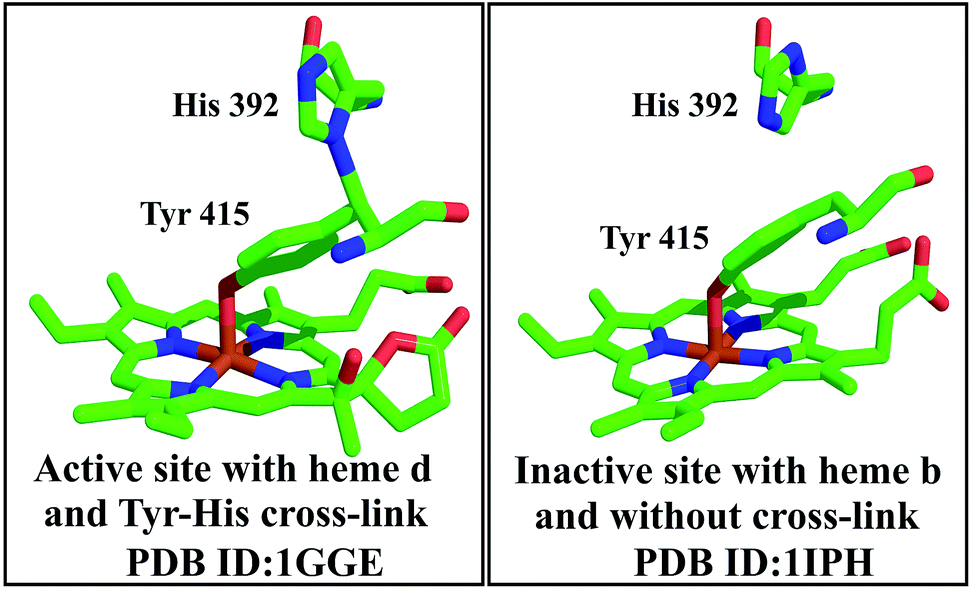 | ||
| Fig. 9 Each subunit of the homotetramer of catalase HP II contains the Tyr–His cross-link. The active site bears the cross-link (left),18 whereas, in the absence of the cross-link (right), the protein is inactive.19 | ||
However, this residue is not conserved in all catalases, and is therefore not essential for all members of this family. For example, the catalase from Proteus mirabilis possesses a methionine sulphone near the active site.18 The proposed mechanism for the conversion, although not satisfactory and requiring revision based on further experimental analysis, is shown in Fig. 10.19a The current model shows that the two reactions, the heme conversion and the novel bond formation, are coupled by the catalase. The mutant forms lacking the ability to convert heme support this mechanism. The linkage may also add rigidity to the active centre and the movement of the electrons can be favoured by the extended structure.19
 | ||
| Fig. 10 Proposed mechanism of formation of the His–Tyr bond in E. coli catalase HP II is shown. Figure re-drawn with permission from Bravo et al.19a | ||
 | ||
| Fig. 11 Crystal structure of the catalase I homodimer shown here from N. crassa.20 The unusual Cys 356–Tyr 379 cross-link formed near the active site is shown on the right. | ||
Oxygenases
The enzyme contains a cofactor in its active site, which contains a thio–ether link between the sulphur of the cysteine and a side chain aromatic carbon of tyrosine (Fig. 12).22 The cofactor is not essential for the catalytic action, but the formation of the cofactor enhances the catalytic action and the catalytic half-life of the enzyme.24 It is also interesting that the cysteine substrate is important for the cofactor formation. When the cysteine levels are low within the cell, the enzyme is degraded by ubiquitination, which helps in conserving the cysteine levels in the cell. When the cysteine levels are high, the cofactor is formed, which increases the catalytic half-life of the enzyme by nearly 10-fold, and helps in clearing the excess cysteine.25 Under extreme conditions of cysteine accumulation within the cell, up to 450-fold enhancement in the enzymatic activity is observed as a consequence of this modification, which helps in effective clearing of the excess cysteine. The proposed mechanism for the cofactor synthesis has been deduced by Stipanuk et al.24 Cysteine dioxygenase is one of the very few examples where a non-disulphide reversible post-translational protein modification is observed.
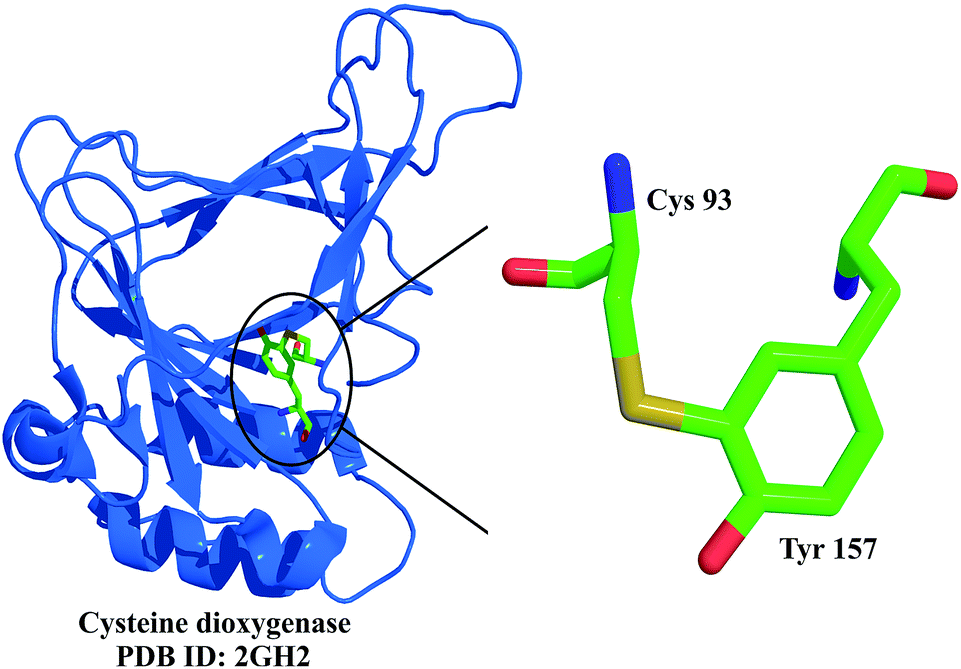 | ||
| Fig. 12 Crystal structure of cysteine dioxygenase, shown here as a ribbon diagram, highlighting the thio–ether link between Cys 93 and Tyr 157.22 | ||
Cross-links with supplementary modifications
Protein PTMs that generate catalytic and redox-active sites usually involve cross-linking coupled with other modifications, primarily side chain oxidation.2d For example, intrinsically formed redox cofactors predominantly involve Tyr or Trp side chains. These quinone modifications have been extensively reviewed recently,26 and are therefore presented here in a succinct manner. Other modifications that involve backbone residues and render the enzyme cofactor-independent are described separately.Quinones
TTQ formation is a result of post-translational modification of two tryptophan residues, in which one or both tryptophans may be oxidized. It is a prevalent cofactor in several amine dehydrogenases.32 For example, in MADH from P. denitrificans, Trp 57 undergoes oxidation at two sites on the indole ring, following which it establishes a covalent link to the indole ring of Trp 108. The protein MauG plays an important role in the synthesis of TTQ,33 as MauG inactivation by site-directed mutagenesis adversely affects TTQ formation.33 MauG particularly participates in the introduction of the second oxygen into the monohydroxylated indole ring of Trp 57 and in covalently linking this ring to Trp 108.34 It appears that MauG and MauL play a concerted role in generating radical intermediates on the pre-MADH substrate. While the cross-linking and oxygen incorporation are directed by the substrate, details of the order of events and the mechanism of MADH modification are yet to be deduced.36
MADH is a heterodimer of two α-subunits (45 kDa each) and two β-subunits (14 kDa each). The β-subunit houses the TTQ (Fig. 13).35 The cofactor is important for the proper assembly of the α- and β-subunits and in its absence dissociation of the subunits occurs. The cofactor is also essential for the redox and catalytic properties of the enzyme, which catalyses the deamination of methylamine to formaldehyde and ammonia (Fig. 14).
 | ||
| Fig. 13 The TTQ cofactor (left) found in the MADH heterodimer (right).35 | ||
 | ||
| Fig. 14 Role of TTQ in the redox reaction catalysed by MADH. Only the quinone region of TTQ is shown, for simplicity of illustration. Figure reproduced with permission from Chen et al.35 | ||
The catalysis proceeds via a reductive reaction, where a Schiff base is formed between the amine substrate and C6 of TTQ.35 Release of the iminoquinone intermediate is accompanied by reduction of the adduct. A general base is required to abstract a proton from the methyl carbon atom, which leads to reduction of the TTQ cofactor. Regeneration of the TTQ cofactor is achieved in the oxidative reaction of the catalysis, where two electrons are transferred from a type-I copper protein (amicyanin in the case of MADH), with the release of the ammonia product and the corresponding aldehyde.37
The P. denitrificans QHNDH α-subunit has 489 residues and is a di-heme c-type cytochrome with four Cys involved in thio–ether linkages to the two heme groups. The β-subunit has a seven-bladed β-propeller motif, folded as a single domain, and comprises 337 residues. The γ-subunit is the smallest (82 residues) and is a globular protein with little secondary structure. It is sandwiched between the α- and β-subunits. Despite the presence of four Cys residues, there are no disulphides in this subunit. The stability of this subunit arises from the three unusual cross-links formed by key residues, namely Cys–Asp and Cys–Glu, for structural stability (discussed later), and cysteine tryptophylquinone (CTQ), shown in Fig. 15, which forms a part of the active site.38,39 These cross-links are formed between the sulphurs of cysteines and either the β- or γ-methylene carbons of aspartic or glutamic acid and provide structural stability to QHNDH,38,39 in a manner similar to the inter-subunit disulphides of MADH.
 | ||
| Fig. 15 The smallest (γ) subunit of QHNDH (shown in pink) contains the CTQ cofactor, formed by the covalent cross-linking of a cysteine side chain with the oxidised indole of tryptophan.38 | ||
The mechanism by which the CTQ modification occurs is poorly understood. It has been shown that the ORF2 protein of P. denitrificans (putative [Fe–S]-cluster and S–Ado–Met (SAM)-binding protein) plays an important role in the post-translational processing of the γ-subunit.40 Furthermore, the α-subunit contains two c-type heme moieties, and it has been speculated that this di-heme subunit may play a role in the biosynthesis of CTQ in a mechanism similar to the role of MauG in TTQ biosynthesis.26a It is, however, evident that the QHNDH and radical SAM enzyme are encoded in the same gene region in P. denitrificans.
CTQ is important for the catalytic activity of QHNDH. The overall reaction catalysed by QHNDH is similar to that with MADH (see Fig. 14).41 Unlike MADH, however, it is difficult to separate the reductive and oxidative half-reactions as the intermediate electron acceptors (the two heme units) are present within the α-subunit of the enzyme. Asp 33 in the γ-subunit serves as the active site base for proton abstraction.41 The re-oxidized QHNDH is generated when these acceptors donate the electrons to azurin in Pseudomonas putida,38 or cytochrome c550 in P. denitrificans.39,42
 | ||
| Fig. 16 Crystal structure of glucose dehydrogenase (shown as a ribbon diagram) revealing the presence of the PQQ cofactor, highlighted here as stick representation.43 | ||
 | ||
| Fig. 17 Proposed function of PQQ in catalysis. Figure reproduced with permission from Elias et al.47 | ||
PQQ is also important for its antioxidant properties and as a growth-promoting factor.48 It has been reported that nanomolar oral consumption of PQQ increases B- and T-cells' responsiveness to mitogens and improves reproductive outcomes and neurological function in rodents.49 PQQ is post-translationally derived from Glu and Tyr amino acids. The process of its biogenesis has not been deduced completely. Based on mutational studies, structural and functional analysis, and sequence homology, it is proposed that PQQ is derived from a precursor peptide PqqA, assisted by several pqqA–F gene products. The mechanism that is currently proposed for the cofactor biosynthesis is presented in detail elsewhere.50
Backbone modifications
Being the oldest enzyme, histidine ammonia lyase (histidase) is one of the best characterized of the three ammonia lyases. In the first step of histidine degradation, catalysed by histidase, the α-amino group is eliminated, resulting in the formation of an α,β-unsaturated trans-urocanate. For this deamination reaction to be carried out, an electrophile is required. X-ray crystallographic studies show that the prosthetic group is an electrophile, 4-methylidene-imidazole-5-one (MIO).51,52 This cofactor, formed by the spontaneous cyclization and dehydration of a tripeptide Ala–Ser–Tyr, is present in all three aromatic amino acid ammonia lyases.51,52 The mechanism of formation of MIO is similar to that of green fluorescent protein (GFP; discussed later).52a
The mechanism by which the degradation is catalysed by these enzymes is shown in Fig. 18. The process involves the electrophilic attack of the histidine side chain on MIO (step 1 in Fig. 18). This is followed by Cβ activation and proton removal by an enzymatic base (step 2). The removal of ammonia in step 3 of the catalytic process now facilitates the regeneration of MIO (and the enzyme active site) along with the release of the deaminated product (step 4).
 | ||
| Fig. 18 Mechanism of MIO action. Figure adapted with permission from Schwede et al.51 Copyright (1999) American Chemical Society. | ||
The challenging catalytic elimination reaction of abstracting the Cβ hydrogen necessitates that the neighbouring highly acidic amino group be maintained in a protonated form, so that it forms a good leaving group. This process requires an electrophilic group to catalyse the reaction. It has been shown by X-ray crystallographic studies, mutational studies and chemical modification experiments that the MIO formed autocatalytically from the Ala–Ser–Tyr triad acts as an excellent electrophilic group; the enhanced electrophilic nature as a result of the cyclization prevents delocalization of the nitrogen lone pairs of Ser and Tyr into the α,β-unsaturated carbonyl system, thereby making the non-oxidative deamination reaction of His, Tyr and Phe feasible.51,52
The catalytic triad formed by backbone cyclization was believed to be dehydroalanine for several decades, till electron cloud density in the X-ray crystallographic map of this protein (2.1 Å resolution) established the structure of MIO (Fig. 19).51 The presence of this modification makes histidase independent of the cofactors (PLP, vitamin B12 and AMP) otherwise observed in other organisms.51 Histidase catalyses the first step in the degradation of histidine to urocanic acid, which undergoes further metabolism to generate glutamate. Malfunctioning of histidases results in a condition called histidinemia.
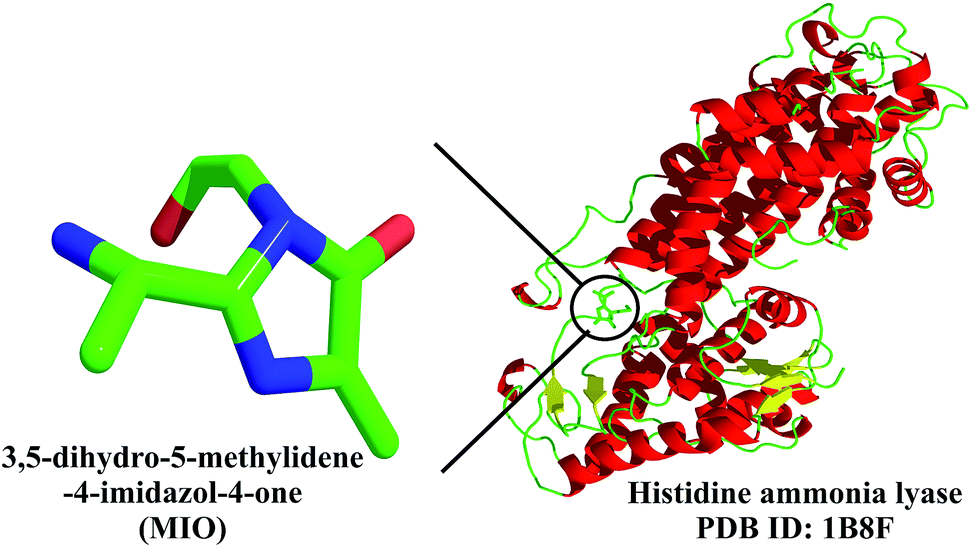 | ||
| Fig. 19 High-resolution crystal structure of histidase, shown here in ribbon form, with the MIO cofactor highlighted.51 | ||
In tyrosine ammonia lyase from yeast Rhodosporidium toruloides, the MIO cofactor is formed by Ala 149, Ser 150 and Gly 151.52c Furthermore, it is speculated that the MIO cofactor carries a covalently attached nucleophilic ammonium adduct. Two spatially proximal Tyr, a Gly and Arg residues are conserved in all the ammonia lyases and provide anchoring interaction with the incoming substrate. Not surprisingly, mutation of these residues dramatically lowers the enzymatic activity of MIOs.52c
Similarly, phenylalanine ammonia lyase resembles histidase, with the exception of an ∼200-residue N-terminal extension in the former, which controls active site access of the substrate.52b In plants, the MIO cofactor of this enzyme is generated by the Ala–Ser–Gly triad autocatalytically, by water elimination.52b It is proposed that this autocatalytic event is driven by mechanical pressure during the refolding process, similar to histidase. Furthermore, the autocatalytic nature of prosthetic group generation in this enzyme (and other lyases) ensures a cofactor-independent upregulation in parsley (Petroselinum crispum).52b
Despite conservation and similarity between the ammonia lyases at the active site, loops positioned at the aromatic cluster of the active site discriminate between the three aromatic substrates and lower cross-reactivity. For instance, it has been shown that in tyrosine ammonia lyase, a His 89 (numbering from R. toruloides) imidazole is involved in distinguishing Tyr from Phe and His.52c These enzymes are actively investigated for their medical relevance in phenylketonuria and other genetic diseases associated with amino acid metabolism.
Surprisingly, histidine decarboxylase isolated from Lactobacillus 30a was found to work independently of PLP. The 2.5 Å crystal structure of this protein revealed that an intrinsic cofactor was generated by the autocatalytic non-hydrolytic serinolysis of Ser 82 of the proenzyme (which becomes Ser 1 of the α-chain of the active enzyme). The pyruvate thus formed from serine forms a Schiff base with the substrate and facilitates the decarboxylation reaction mediated by PLP in a PLP-independent manner.1 S-Adenosylmethionine decarboxylase (AdoMetDC), involved in polyamine biosynthesis in bacteria, plants and humans, also undergoes internal post-translational serinolysis, generating the pyruvoyl moiety for decarboxylation. This has been reviewed extensively elsewhere.53
The enzyme is regulated by cellular hydrogen peroxide levels; the latter is required for numerous signal transduction pathways, particularly those mediated by tyrosine kinases. H2O2 acts by transiently inhibiting PTPs by converting the Cys–SH to Cys–S–OH. Indeed, different oxidation states of the catalytic cysteine residue (sulphenic, sulphinic and sulphonic acid derivatives) have been observed in the crystal structure.54 Some of these oxidation states, formed in the presence of excess H2O2, can irreversibly inhibit the enzymatic activity.
How is irreversible inhibition prevented in the cell? The answer to this question came with the crystal structure of PTP 1B, with unusual electron density close to the side chain of the catalytic cysteine. A bond length of 1.7 Å, measured between Sγ of Cys 215 and the backbone nitrogen of Ser 216,54a,b could only be explained by the formation of a five-membered puckered ring containing a S–N covalent bond (Fig. 20). It has been proposed that the sulphenyl-amide bond is generated by the nucleophilic attack of the amide on the sulphenic acid form of the cysteine side chain. The cysteine is regenerated by another nucleophilic attack on the S–N bond.54a,b
 | ||
| Fig. 20 Five-membered puckered ring formed by a 1.7 Å sulphenyl-amide covalent bond formed between the Sγ of Cys 215 and the backbone nitrogen of Ser 216 in protein tyrosine phosphatase 1B.54 | ||
It has been proposed that the sulphenyl-amide intermediate is an elegant protection mechanism of the catalytic cysteine residue (pKa ∼ 5.4) from irreversible oxidation.54a,b The associated conformational changes due to the backbone constraint arising from the puckered five-membered ring can signal an inactive state of the enzyme. Furthermore, this sulphenyl-amide state of the protein can readily be restored, allowing for the reactivation of PTP 1B.
Cross-links in non-enzymatic proteins
Cross-linking between side chains, side chain and backbone and other backbone modifications, are also observed in a small, yet significant, class of non-enzymatic proteins. Some of these modifications, such as a Tyr–Tyr cross-link in bovine γB crystallin, forms due to ageing, whereas a similar Tyr–Tyr cross-link in extrusin from tomato cell wall confers rigidity on the structure. This section highlights some of the known and interesting examples identified in structural proteins, which may provide additional unique characteristics to the protein molecule. A small note on unique PTMs observed in peptides has also been included.Green fluorescent protein (GFP)
The ∼27 kDa green fluorescent protein, known popularly as GFP, has been one of the most prevalent and widely used proteins for reporter-based gene expression, protein localization tags, biosensors, and several other applications in bacteria, yeast and higher organisms, including mammalian systems, since its first application in ∼1992 by Douglas Prasher.56 This protein was first identified and isolated from the jellyfish Aequorea victoria,57 and is an 11-stranded barrel with six short helical structures. Despite widespread applications of GFP, the term 4-(p-hydroxybenzylidene)-imidazolidin-5-one (HBI) is seldom known in the GFP user community. The distinctive fluorescent nature of GFP is due to this unique chromophore HBI (Fig. 21), formed at the centre of the barrel between residues Ser/Thr 65–Tyr 66–Gly 67 when GFP is correctly folded. Detailed mechanistic examination of this protein has been possible after the crystal structure was solved,55 which has also serendipitously led to the development of several other fluorescent analogues of GFP by modulating the HBI chromophore within the barrel. Our current understanding of this protein, as well as its derivatives, is a result of extensive work carried out on GFP and its applications by several scientists including Osamu Shimomura, Martin Chalfie and Roger Y. Tsien, which earned them the Nobel Prize in Chemistry in the year 2008.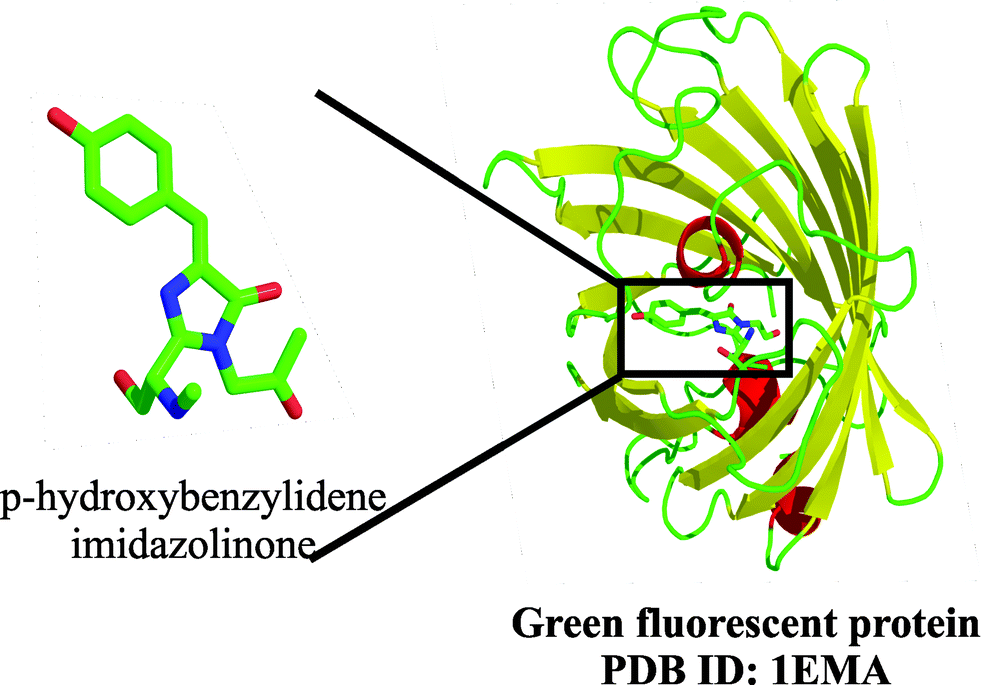 | ||
| Fig. 21 Ribbon diagram of GFP, highlighting the chromophore generated by backbone cyclization, in stick form.55 | ||
Information on the application of GFP and its analogues is extensively described in several reports; the formation of the HBI chromophore itself is discussed here. The fluorescent nature of the chromophore arises due to an unusual covalent bond formed by backbone cyclization of residues Ser 65 (backbone carboxyl group) and Gly 67 (backbone amide) followed by dehydration leading to the formation of the imidazolinone. The final oxidation reaction of the Tyr 66 Cα–Cβ bond that uses molecular oxygen conjugates the phenolic ring with the imidazolinone. The phenolate ion thus generated serves as the light emitter. Presence of an unusual post-translational protein modification leading to chromophore formation was first identified by papain cleavage of GFP. Characterization of the hexapeptide F64-Q69, which was generated during this process, led to identification of this unusual cyclization. The peptide, in isolation, is non-fluorescent, and requires the local environment generated by the folded protein to exhibit green fluorescence.
In native GFP from A. victoria, the blue-green light is emitted at a maximum wavelength of ∼508 nm;58 the emission maximum can be modified based on the chemical nature of the residues involved in the conjugation and chromophore formation.59 This extensive conjugation not only confers the chromophoric property on the protein but also shows considerable thermal stability after formation. The chromophore formation is an autocatalytic event and occurs in the absence of cofactors;55a the only requirements for complete cyclization are a proper folded conformation of the protein as well as temperatures of 30 °C or above.2b Additionally, isomerization across the exo-methylene bond is inhibited by its geometry; this results in a highly efficient green light emission by minimizing non-radiative pathways for decay of the excited fluorophore. The mechanism of the biogenesis of the chromophore is described by Tsien et al.2a
Ranasmurfin
The formation of stable chromophores using unusual post-translational modifications is not restricted to GFP. A blue-green protein identified in the foam nests of the Malaysian tree frog is one of several proteins and macromolecules that confer stability and adhesion properties on the nest. This blue-green protein, identified as ranasmurfin, is a 13 kDa homodimer, with each subunit containing three disulphides and a covalent lysyl tyrosyl quinone linkage (LTQ) established between Tyr 2 and Lys 31, as well as Lys 30 and Tyr 108 (Fig. 22), in each subunit.60 The Lys 30–Tyr 108 LTQ is additionally modified by a nitrogen atom to link the two subunits and give rise to a bis-LTQ linkage (a four-residue Lys–Tyr–N–Tyr–Lys link), which now covalently links the dimer. This bis-LTQ, along with two histidines, forms a coordinate bond with zinc. | ||
| Fig. 22 The unusual blue protein found in the Malaysian frog nest, ranasmurfin, contains two subunits, each of which bears the unusual modification, LTQ, between Lys 31 and Tyr 2 (top, left). The bis-LTQ linkage is connected by an atom, speculated to be nitrogen (top, right). The ribbon diagram of ranasmurfin is provided in the bottom.60b | ||
While the LTQ modification is observed in other proteins (discussed later), the extended aromatic system, along with the metal coordination, gives rise to π-cloud redistribution across the amide–aromatic bonds, and thereby confers the characteristic blue chromophore on ranasmurfin, with an absorbance range of 500–700 nm and emission maxima at 680–700 nm. The chromophore has two ionisable groups with pKa values of 6.0 and 9.0, which make it exhibit reversible spectral shifts with changes in pH. The blue colour is also unaffected by reducing agents such as NaBH4; however, the colour slowly fades on treatment with EDTA and DTT, and rapid bleaching occurs in the presence of NBS.60b While the mechanism by which the LTQ post-translational modification occurs and its biological importance is unknown, the cross-link confers high inter-subunit stability on the protein and resistance to partial proteolysis and denaturants, and it is speculated that the bis-LTQ plays a role in the mechanical adhesion properties of the foam.60b
Collagen
The basic components of the basement membranes that underlie the epithelia of all metazoans are collagen IV networks. They are formed from a family of six polypeptide chains (α1–α6). These associate into three different subtypes of protomers (α1α1α2, α3α4α5, and α5α5α6), which are triple-helical in nature. Self-assembly of the protomer occurs through end-to-end associations, where four protomers associate tail-to-tail through their amino termini. The carboxyl termini of two protomers, on the other hand, associate head-to-head through the non-collagenous (NC1) domains and give rise to dimers. The trimeric non-collagenous NC1 domains therefore exist as hexamers at the interface of the head-to-head interactions (Fig. 23).61 | ||
| Fig. 23 (Top) Crystal structures of collagen IV, highlighting the absence (left)64,65 and presence (right)63 of the cross-link at the oligomerization interface. Lys 211–Met 93 interaction is represented here as spheres. The PDB IDs and resolutions are indicated alongside the respective structures. (Bottom) The six unusual cross-links identified in the 1.9 Å crystal structure are highlighted. The protein is shown as a ribbon diagram and residues involved in cross-linking are shown as sticks.63 | ||
The NC1 domains of three monomer chains interact to form a trimer, which nucleates the formation of a triple helix. NC1 trimers from two protomers in the network assembly interact to form a hexameric structure. Exposure to acidic pH or denaturants drives dissociation of the hexameric assembly, yielding monomers and dimers. The existence of dimers in the denaturing conditions is because of the presence of cross-link(s) at the trimer–trimer interface.62 These cross-links are present between the α1-like monomers: α1–α1, α1–α5, and α3–α5 and α2-like monomers: α2–α2, α2–α6, and α4–α4.61,62
It has been shown chemically, and by using X-ray crystallography (1.9 Å resolution), that the cross-link site of the trimer networks contains six unusual covalent thio–ether bonds between Met and Lys (Met 93 and Lys 211 in the case of human placenta collagen IV), formed through post-translational modifications (Fig. 23).63 There are two post-translational events believed to be involved in this novel bond formation. Firstly, Lys is hydroxylated to Hyl (hydroxylysine) within the non-collagenous domain, which is followed by formation of the link between Hyl 211 and Met 93 of two trimeric non-collagenous domains.61 The exact mechanism by which these post-translational modifications occur is still unknown.
Surprisingly, X-ray crystallographic structure, solved to a resolution of 1.5 Å, did not show any supporting evidence for the existence of this cross-link (Fig. 23).64 On the contrary, mass spectrometric analysis, Edman degradation and amino acid analysis that were carried out using tryptic digests of the monomer and dimer subunits, revealed the existence of a novel cross-link between Hyl 211 and Met 93.61 However, it is acknowledged that these covalent linkages confer the necessary structural stability for oligomerization, since monomers of the non-collagenous domains which lack the cross-link are involved in Goodpasture disease and Alport syndrome.63,66
Hemoglobin O
Truncated hemoglobins (trHb) are hemoproteins observed in microorganisms and are believed to be involved in cell respiration, particularly at the stationary phase of cell growth. The crystal structure of trHbO, a dodecamer encoded by the glbO gene in Mycobacterium tuberculosis, revealed an unusual cross-link between Tyr 36 Cε2 and the neighbouring Tyr 23 O (1.43 Å distance), which gives rise to rigidity between the two orthogonal aromatic rings. The cross-link positions the oxygen of Tyr 36 in the necessary geometry for stabilizing the heme-bound cyanide.67 The ether link is speculated to be formed in solution before dodecamer assembly through an unknown oxidative mechanism; its functional significance, besides establishing the necessary Tyr geometry, is still unclear.67Peptides and toxins
Several examples of ribosomally and non-ribosomally synthesized peptides are known to bear unusual post-translational modifications. The unique protease resistance and structural stability conferred by these cross-links are critical for the function of these peptides. For example, bacteriocins, the antimicrobial bacterial peptides, are cationic, ribosomally synthesized, and 25–60 residues in length, and several of these molecules are extensively modified. For instance, nisin A from Lactococcus lactis, a food preservative and broad-spectrum bacteriocin, is 34 amino acids long and contains three unusual amino acids and five thio–ether bridges.68 Similarly, subtilosin A, a Bacillus subtilis antimicrobial peptide, bears three S–C links between Cys and Phe/Thr, in addition to backbone cyclization.69 Phalloidin, produced by the fungus Amanita phalloides, is another bicyclic heptapeptide with a transannular thio–ether bridge connecting cysteine and tryptophan side chains.70 The biosynthesis and unusual chemical properties of the ∼20 distinct classes of PTM peptides is described in a recent review by van der Donk et al.2iConclusions
Crystallographic and biochemical studies have unearthed novel post-translational modifications within enzyme active sites that have structural and functional diversity. These modifications mainly serve as cofactors, making these enzymes independent of the usual cofactors found in proteins. Most of the enzymes bearing modifications carry out redox reactions, and usually involve a metal-dependent redox reaction in the establishment of the cross-link. The unusual post-translational protein modifications described in this review have remained alien for long, despite their evolutionary conservation, with only a few studies that report the chemistry behind their synthesis and their mode of action at the molecular level.The advent of biophysical techniques has helped unravel some of these modifications, many of which continue to remain serendipitous discoveries. In the recent review by Klinman on quinone cofactors, she has commented on the evolutionary redundancy in enzymes that catalyse similar reactions.26b Furthermore, it is evident from our categorization of the unusual PTMs identified in this review, that a vast majority of these modifications are predominant in oxido-reductive (redox) reactions and in structural proteins. Cofactors for redox cycles are evolutionarily chosen for their ability to act as both electron donors and acceptors at different stages of the reaction, due to their extensively conjugated π-electron network. Unusual PTMs seem to cater to the requirements of redox reactions, suggesting that more examples of such non-disulphide modifications can be obtained from such enzymes.
Redox reactions in proteins are often associated with the generation of free radicals; it is tempting to speculate that these unusual cross-links could have evolved due to accidental modifications at the protein active site by such radicals. Irrespective of their origin, one can safely assume that a larger collection of unusual PTMs will be unearthed in the oxidoreductase family of enzymes. Could these cross-links also perform alternative roles in protecting the enzyme active site from redox damage? As many cross-links show reversibility during catalysis in the protein, it would be of interest to examine their role in the regulation of protein activity under oxidative stress in the host cell, as well as their possible role in cell signalling.54b
Another major property of such PTMs is the associated rigidity they confer on proteins, especially at oligomerization interfaces. Hence one other possible family of proteins that would possess such modifications would include the structural proteins. It would be possible to discover such PTMs using high-resolution crystallography, coupled with mass spectrometric mapping, as employed earlier for PTP 1B,54b if such proteins are actively explored for unusual modifications. Surprisingly, however, in the cases of GFP, ranasmurfin and other unusual chromophores, it has been speculated that these molecules evolved via related autocatalytic mechanisms by simple residue substitutions at the modification site to achieve the diverse chromophores.71 Previous studies have also speculated that such modifications were retained by natural selection, due to their role in alternative, hitherto unknown, functions.71 It would be of interest to identify such functions and develop synthetic mimetics that have regulatory roles in key biochemical processes.
The range of methodologies currently available for in vitro synthesis of such unusual cross-links does not span all the combinations generated in nature between the various amino acids. Nevertheless, the few strategies that have indeed been successfully developed to obtain di-tryptophan and di-tyrosine cross-links72 in small peptides and proteins not only have potential applications in the generation of artificial enzymes, but also serve to pave the way for more complex chemistries for the production of many other unusual modifications.
Deducing the mechanism of formation of such unusual cross-links between amino acid side chains in vivo would facilitate the development of reagents that would allow their generation in in vitro systems. The scientific community has now acknowledged that non-disulphide cross-links are also possible in proteins and the observation of unusual or anomalous electron density in protein crystal structures, for example, need not be artefacts of data collection. Such cross-links, in addition to being of extreme interest themselves, can be exploited in the design and chemical synthesis of small peptide molecules that can be engineered to carry out protein-like reactions. Such catalytic peptides are of great scope in both industry and in curing diseases caused by enzyme deficiencies.
There is escalating interest in the application of these cross-links to the generation of artificial enzymes and synthetic scaffolds with cofactor-independent catalytic activity. By mimicking the algorithms followed by nature in bringing about these modifications, it is possible to design strategies in drug targeting and create modified enzymes with a minimal effect on the geometry of the active site, which can perform catalytic activity devoid of cofactors and also provide extra structural stability to the proteins. It would therefore come as no surprise if many more such examples are uncovered or generated over the years, in a wider repertoire of proteins and peptides that will eventually give rise to a massive number of such unusual post-translational modifications, and will no longer leave them considered unusual.
Acknowledgements
B.R. is supported by the INSPIRE fellowship from IISER Bhopal. R.M. is a recipient of the Ramalingaswami fellowship from the Department of Biotechnology, Govt. of India. This work is supported by intramural funds.Notes and references
- T. Gallagher, D. A. Rozwarski, S. R. Ernst and M. L. Hackert, J. Mol. Biol., 1993, 230, 516–528 CrossRef CAS PubMed.
- (a) A. B. Cubitt, R. Heim, S. R. Adams, A. E. Boyd, L. A. Gross and R. Y. Tsien, Trends Biochem. Sci., 1995, 20, 448–455 CrossRef CAS PubMed; (b) G. N. Phillips, Jr, Curr. Opin. Struct. Biol., 1997, 7, 821–827 CrossRef; (c) N. M. Okeley and W. A. van der Donk, Chem. Biol., 2000, 7, R159–R171 CrossRef CAS PubMed; (d) J. P. Klinman, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 14766–14768 CrossRef CAS PubMed; (e) L. Xie and W. A. van der Donk, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 12863–12865 CrossRef CAS PubMed; (f) J. W. Whittaker, Arch. Biochem. Biophys., 2005, 433, 227–239 CrossRef CAS PubMed; (g) X. B. Zhao, J. Suarez, A. Khajo, S. W. Yu, L. Metlitsky and R. S. Magliozzo, J. Am. Chem. Soc., 2010, 132, 8268–8269 CrossRef CAS PubMed; (h) D. Rokhsana, A. E. Howells, D. M. Dooley and R. K. Szilagyi, Inorg. Chem., 2012, 51, 3513–3524 CrossRef CAS PubMed; (i) P. G. Arnison, M. J. Bibb, G. Bierbaum, A. A. Bowers, T. S. Bugni, G. Bulaj, J. A. Camarero, D. J. Campopiano, G. L. Challis, J. Clardy, P. D. Cotter, D. J. Craik, M. Dawson, E. Dittmann, S. Donadio, P. C. Dorrestein, K. D. Entian, M. A. Fischbach, J. S. Garavelli, U. Goransson, C. W. Gruber, D. H. Haft, T. K. Hemscheidt, C. Hertweck, C. Hill, A. R. Horswill, M. Jaspars, W. L. Kelly, J. P. Klinman, O. P. Kuipers, A. J. Link, W. Liu, M. A. Marahiel, D. A. Mitchell, G. N. Moll, B. S. Moore, R. Muller, S. K. Nair, I. F. Nes, G. E. Norris, B. M. Olivera, H. Onaka, M. L. Patchett, J. Piel, M. J. Reaney, S. Rebuffat, R. P. Ross, H. G. Sahl, E. W. Schmidt, M. E. Selsted, K. Severinov, B. Shen, K. Sivonen, L. Smith, T. Stein, R. D. Sussmuth, J. R. Tagg, G. L. Tang, A. W. Truman, J. C. Vederas, C. T. Walsh, J. D. Walton, S. C. Wenzel, J. M. Willey and W. A. van der Donk, Nat. Prod. Rep., 2013, 30, 108–160 RSC.
- F. Jordan and M. S. Patel, Thiamine: Catalytic Mechanisms in Normal and Disease States (Oxidative Stress and Disease), CRC Press, 2003 Search PubMed.
- (a) N. Ito, S. E. Phillips, C. Stevens, Z. B. Ogel, M. J. McPherson, J. N. Keen, K. D. Yadav and P. F. Knowles, Nature, 1991, 350, 87–90 CrossRef CAS; (b) S. J. Firbank, M. S. Rogers, C. M. Wilmot, D. M. Dooley, M. A. Halcrow, P. F. Knowles, M. J. McPherson and S. E. Phillips, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 12932–12937 CrossRef CAS.
- S. Yoshikawa, K. Shinzawa-Itoh, R. Nakashima, R. Yaono, E. Yamashita, N. Inoue, M. Yao, M. J. Fei, C. P. Libeu, T. Mizushima, H. Yamaguchi, T. Tomizaki and T. Tsukihara, Science, 1998, 280, 1723–1729 CrossRef CAS PubMed.
- D. A. Pratt, R. P. Pesavento and W. A. van der Donk, Org. Lett., 2005, 7, 2735–2738 CrossRef CAS PubMed.
- C. Ostermeier, A. Harrenga, U. Ermler and H. Michel, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 10547–10553 CrossRef CAS.
- G. Buse, T. Soulimane, M. Dewor, H. E. Meyer and M. Bluggel, Protein Sci., 1999, 8, 985–990 CrossRef CAS PubMed.
- M. A. Sharpe and S. Ferguson-Miller, J. Bioenerg. Biomembr., 2008, 40, 541–549 CrossRef CAS PubMed.
- T. Klabunde, C. Eicken, J. C. Sacchettini and B. Krebs, Nat. Struct. Biol., 1998, 5, 1084–1090 CrossRef CAS PubMed.
- E. I. Solomon, U. M. Sundaram and T. E. Machonkin, Chem. Rev., 1996, 96, 2563–2606 CrossRef CAS PubMed.
- C. Eicken, B. Krebs and J. C. Sacchettini, Curr. Opin. Struct. Biol., 1999, 9, 677–683 CrossRef CAS PubMed.
- T. Bertrand, N. A. Eady, J. N. Jones, Jesmin, J. M. Nagy, B. Jamart-Gregoire, E. L. Raven and K. A. Brown, J. Biol. Chem., 2004, 279, 38991–38999 CrossRef CAS PubMed.
- (a) Y. Zhang, B. Heym, B. Allen, D. Young and S. Cole, Nature, 1992, 358, 591–593 CrossRef CAS PubMed; (b) R. A. Ghiladi, G. M. Knudsen, K. F. Medzihradszky and P. R. Ortiz de Montellano, J. Biol. Chem., 2005, 280, 22651–22663 CrossRef CAS PubMed.
- B. Bhaskar, C. E. Immoos, H. Shimizu, F. Sulc, P. J. Farmer and T. L. Poulos, J. Mol. Biol., 2003, 328, 157–166 CrossRef CAS PubMed.
- E. J. Gilbert, J. D. Chisholm and D. L. Van Vranken, J. Org. Chem., 1999, 64, 5670–5676 CrossRef CAS PubMed.
- P. C. Loewen, J. Switala, I. von Ossowski, A. Hillar, A. Christie, B. Tattrie and P. Nicholls, Biochemistry, 1993, 32, 10159–10164 CrossRef CAS.
- W. Meilk-Adamyan, J. Bravo, X. Carpena, J. Switala, M. Mate, I. Fita and P. C. Loewen, Proteins: Struct., Funct., Genet., 2001, 44, 270–281 CrossRef PubMed.
- (a) J. Bravo, I. Fita, J. C. Ferrer, W. Ens, A. Hillar, J. Switala and P. C. Loewen, Protein Sci., 1997, 6, 1016–1023 CrossRef CAS; (b) J. Bravo, N. Verdaguer, J. Tormo, C. Betzel, J. Switala, P. C. Loewen and I. Fita, Structure, 1995, 3, 491–502 CrossRef CAS PubMed.
- A. Diaz, E. Horjales, E. Rudino-Pinera, R. Arreola and W. Hansberg, J. Mol. Biol., 2004, 342, 971–985 CrossRef CAS PubMed.
- (a) G. S. Jacob and W. H. Orme-Johnson, Biochemistry, 1979, 18, 2967–2975 CrossRef CAS PubMed; (b) G. S. Jacob and W. H. Orme-Johnson, Biochemistry, 1979, 18, 2975–2980 CrossRef CAS PubMed.
- C. R. Simmons, Q. Liu, Q. Huang, Q. Hao, T. P. Begley, P. A. Karplus and M. H. Stipanuk, J. Biol. Chem., 2006, 281, 18723–18733 CrossRef CAS PubMed.
- J. E. Dominy, Jr, J. Hwang and M. H. Stipanuk, Am. J. Physiol.: Endocrinol. Metab., 2007, 293, E62–E69 CrossRef PubMed.
- J. E. Dominy, J. Hwang, S. Guo, L. L. Hirschberger, S. Zhang and M. H. Stipanuk, J. Biol. Chem., 2008, 283, 12188–12201 CrossRef CAS PubMed.
- M. H. Stipanuk, I. Ueki, J. E. Dominy, Jr, C. R. Simmons and L. L. Hirschberger, Amino Acids, 2009, 37, 55–63 CrossRef CAS PubMed.
- (a) V. L. Davidson, Mol. BioSyst., 2011, 7, 29–37 RSC; (b) J. P. Klinman and F. Bonnot, Chem. Rev., 2014, 114, 4343–4365 CrossRef CAS PubMed.
- J. A. Bollinger, D. E. Brown and D. M. Dooley, Biochemistry, 2005, 44, 11708–11714 CrossRef CAS PubMed.
- H. M. Kagan and W. Li, J. Cell. Biochem., 2003, 88, 660–672 CrossRef CAS PubMed.
- Q. Xiao and G. Ge, Cancer Microenviron., 2012, 5, 261–273 CrossRef CAS PubMed.
- J. Wu, C. Cai, D. N. A. Tong and H. C. Hou, Genet. Test. Mol. Biomarkers, 2012, 16, 915–919 CrossRef CAS PubMed.
- S. A. Salisbury, H. S. Forrest, W. B. Cruse and O. Kennard, Nature, 1979, 280, 843–844 CrossRef CAS.
- W. S. Mcintire, D. E. Wemmer, A. Chistoserdov and M. E. Lidstrom, Science, 1991, 252, 817–824 CrossRef CAS PubMed.
- A. R. Pearson, T. De la Mora-Rey, M. E. Graichen, Y. T. Wang, L. H. Jones, S. Marimanikkupam, S. A. Agger, P. A. Grimsrud, V. L. Davidson and C. M. Wilmot, Biochemistry, 2004, 43, 5494–5502 CrossRef CAS PubMed.
- L. M. R. Jensen, R. Sanishvili, V. L. Davidson and C. M. Wilmot, Science, 2010, 327, 1392–1394 CrossRef CAS.
- L. Chen, M. Doi, R. C. Durley, A. Y. Chistoserdov, M. E. Lidstrom, V. L. Davidson and F. S. Mathews, J. Mol. Biol., 1998, 276, 131–149 CrossRef CAS PubMed.
- V. L. Davidson and A. Liu, Biochim. Biophys. Acta, 2012, 1824, 1299–1305 CrossRef CAS PubMed.
- Z. Zhu and V. L. Davidson, Biochemistry, 1999, 38, 4862–4867 CrossRef CAS PubMed.
- S. Datta, Y. Mori, K. Takagi, K. Kawaguchi, Z. W. Chen, T. Okajima, S. Kuroda, T. Ikeda, K. Kano, K. Tanizawa and F. S. Mathews, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 14268–14273 CrossRef CAS PubMed.
- S. Datta, T. Ikeda, K. Kano and F. S. Mathews, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2003, 59, 1551–1556 CrossRef.
- K. Ono, T. Okajima, M. Tani, S. Kuroda, D. Sun, V. L. Davidson and K. Tanizawa, J. Biol. Chem., 2006, 281, 13672–13684 CrossRef CAS PubMed.
- A. Satoh, O. Adachi, K. Tanizawa and K. Hirotsu, Biochim. Biophys. Acta, 2003, 1647, 272–277 CrossRef CAS.
- K. Takagi, K. Yamamoto, K. Kano and T. Ikeda, Eur. J. Biochem., 2001, 268, 470–476 CrossRef CAS PubMed.
- A. Oubrie, H. J. Rozeboom, K. H. Kalk, A. J. Olsthoorn, J. A. Duine and B. W. Dijkstra, EMBO J., 1999, 18, 5187–5194 CrossRef CAS PubMed.
- C. Anthony, Antioxid. Redox Signaling, 2001, 3, 757–774 CrossRef CAS PubMed.
- J. Park and J. E. Churchich, BioFactors, 1992, 3, 257–260 CAS.
- C. Anthony, Biochem. J., 1996, 320(Pt 3), 697–711 CrossRef CAS PubMed.
- M. D. Elias, M. Tanaka, H. Izu, K. Matsushita, O. Adachi and M. Yamada, J. Biol. Chem., 2000, 275, 7321–7326 CrossRef CAS PubMed.
- K. He, H. Nukada, T. Urakami and M. P. Murphy, Biochem. Pharmacol., 2003, 65, 67–74 CrossRef CAS PubMed.
- T. E. Stites, A. E. Mitchell and R. B. Rucker, J. Nutr., 2000, 130, 719–727 CAS.
- S. Puehringer, M. Metlitzky and R. Schwarzenbacher, BMC Biochem., 2008, 9, 8 CrossRef PubMed.
- T. F. Schwede, J. Retey and G. E. Schulz, Biochemistry, 1999, 38, 5355–5361 CrossRef CAS PubMed.
- (a) M. Baedeker and G. E. Schulz, Structure, 2002, 10, 61–67 CrossRef CAS PubMed; (b) H. Ritter and G. E. Schulz, Plant Cell, 2004, 16, 3426–3436 CrossRef CAS PubMed; (c) G. V. Louie, M. E. Bowman, M. C. Moffitt, T. J. Baiga, B. S. Moore and J. P. Noel, Chem. Biol., 2006, 13, 1327–1338 CrossRef CAS PubMed.
- S. Bale and S. E. Ealick, Amino Acids, 2011, 38, 451–460 CrossRef PubMed.
- (a) A. D. Pannifer, A. J. Flint, N. K. Tonks and D. Barford, J. Biol. Chem., 1998, 273, 10454–10462 CrossRef CAS PubMed; (b) A. Salmeen, J. N. Andersen, M. P. Myers, T. C. Meng, J. A. Hinks, N. K. Tonks and D. Barford, Nature, 2003, 423, 769–773 CrossRef CAS PubMed; (c) R. L. van Montfort, M. Congreve, D. Tisi, R. Carr and H. Jhoti, Nature, 2003, 423, 773–777 CrossRef CAS PubMed.
- (a) F. Yang, L. G. Moss and G. N. Phillips, Jr, Nat. Biotechnol., 1996, 14, 1246–1251 CrossRef CAS PubMed; (b) M. Ormo, A. B. Cubitt, K. Kallio, L. A. Gross, R. Y. Tsien and S. J. Remington, Science, 1996, 273, 1392–1395 CAS.
- D. C. Prasher, V. K. Eckenrode, W. W. Ward, F. G. Prendergast and M. J. Cormier, Gene, 1992, 111, 229–233 CrossRef CAS PubMed.
- O. Shimomura, F. H. Johnson and Y. Saiga, J. Cell. Comp. Physiol., 1962, 59, 223–239 CrossRef CAS PubMed.
- K. Brejc, T. K. Sixma, P. A. Kitts, S. R. Kain, R. Y. Tsien, M. Ormo and S. J. Remington, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 2306–2311 CrossRef CAS.
- D. P. Barondeau, C. D. Putnam, C. J. Kassmann, J. A. Tainer and E. D. Getzoff, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 12111–12116 CrossRef CAS PubMed.
- (a) S. A. McMahon, M. A. Walsh, R. T. Ching, L. G. Carter, M. Dorward, K. A. Johnson, H. Liu, M. Oke, C. Bloch, Jr, M. W. Kennedy, A. A. Latiff, A. Cooper, G. L. Taylor, M. F. White and J. H. Naismith, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2006, 62, 1124–1126 CrossRef CAS PubMed; (b) M. Oke, R. T. Ching, L. G. Carter, K. A. Johnson, H. Liu, S. A. McMahon, M. F. White, C. Bloch, Jr, C. H. Botting, M. A. Walsh, A. A. Latiff, M. W. Kennedy, A. Cooper and J. H. Naismith, Angew. Chem., Int. Ed. Engl., 2008, 47, 7853–7856 CrossRef CAS PubMed.
- R. M. Vanacore, D. B. Friedman, A. J. Ham, M. Sundaramoorthy and B. G. Hudson, J. Biol. Chem., 2005, 280, 29300–29310 CrossRef CAS PubMed.
- R. Vanacore, A. J. Ham, M. Voehler, C. R. Sanders, T. P. Conrads, T. D. Veenstra, K. B. Sharpless, P. E. Dawson and B. G. Hudson, Science, 2009, 325, 1230–1234 CrossRef CAS PubMed.
- M. E. Than, S. Henrich, R. Huber, A. Ries, K. Mann, K. Kuhn, R. Timpl, G. P. Bourenkov, H. D. Bartunik and W. Bode, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 6607–6612 CrossRef CAS PubMed.
- R. M. Vanacore, S. Shanmugasundararaj, D. B. Friedman, O. Bondar, B. G. Hudson and M. Sundaramoorthy, J. Biol. Chem., 2004, 279, 44723–44730 CrossRef CAS PubMed.
- M. Sundaramoorthy, M. Meiyappan, P. Todd and B. G. Hudson, J. Biol. Chem., 2002, 277, 31142–31153 CrossRef CAS.
- R. M. Vanacore, A. J. Ham, J. P. Cartailler, M. Sundaramoorthy, P. Todd, V. Pedchenko, Y. Sado, D. B. Borza and B. G. Hudson, J. Biol. Chem., 2008, 283, 22737–22748 CrossRef CAS PubMed.
- M. Milani, P. Y. Savard, H. Ouellet, P. Ascenzi, M. Guertin and M. Bolognesi, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 5766–5771 CrossRef CAS PubMed.
- A. Guder, I. Wiedemann and H. G. Sahl, Biopolymers, 2000, 55, 62–73 CrossRef CAS PubMed.
- K. Kawulka, T. Sprules, R. T. McKay, P. Mercier, C. M. Diaper, P. Zuber and J. C. Vederas, J. Am. Chem. Soc., 2003, 125, 4726–4727 CrossRef CAS PubMed.
- G. Zanotti, L. Falcigno, M. Saviano, G. D'Auria, B. M. Bruno, T. Campanile and L. Paolillo, Chemistry, 2001, 7, 1479–1485 CrossRef CAS PubMed.
- Y. A. Labas, N. G. Gurskaya, Y. G. Yanushevich, A. F. Fradkov, K. A. Lukyanov, S. A. Lukyanov and M. V. Matz, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4256–4261 CrossRef CAS PubMed.
- (a) E. D. Horowitz, M. G. Finn and A. Asokan, ACS Chem. Biol., 2012, 7, 1059–1066 CrossRef CAS PubMed; (b) J. H. Matthews, T. D. Dinh, P. Tivitmahaisoon, J. W. Ziller and D. L. Van Vranken, Chem. Biol., 2001, 8, 1071–1079 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |