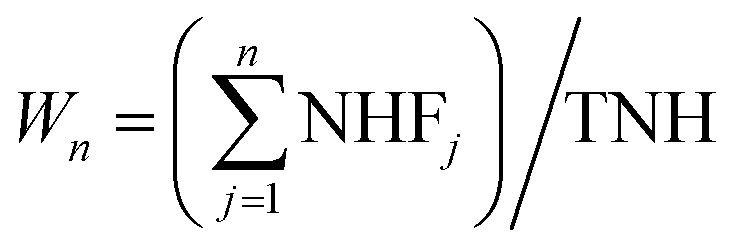DOI:
10.1039/C4RA04642K
(Paper)
RSC Adv., 2014,
4, 36325-36335
OneG-Vali: a computational tool for detecting, estimating and validating cryptic intermediates of proteins under native conditions†
Received
17th May 2014
, Accepted 6th August 2014
First published on 6th August 2014
Abstract
Understanding structural excursions of proteins under native conditions at residue level resolutions is crucial to map energy landscapes of proteins and also to solve the ‘Levinthal paradox’ of protein folding. Native-state hydrogen–deuterium (NS H/D) exchange methods are powerful to structurally characterize cryptic intermediates (CIs) populating sparsely in the unfolding kinetics of proteins under conditions favoring folded conformations of the proteins. However, the methods are not applicable to proteins that are susceptible to denaturation or degradation or aggregation in the course of exchange experiments and also to proteins that are smaller in size (<10 kDa) in general. We have herein demonstrated a novel computational tool, OneG-Vali, which predicts the possible existence of cryptic intermediates of proteins in a qualitative and quantitative manner. And, the tool validates the prediction efficiency by comparing multistate unfolding curves defined by the tool with pseudo two-state unfolding curves of the proteins determined by macroscopic methods. In addition, the OneG-Vali facilitates accounting for the effect of cis–trans proline isomerization on estimating the population of CIs defined by the tool and also by experimental methods. The prediction accuracy of the tool is validated using proteins such as cytochrome c, apocytochrome b562, the third domain of PDZ and T4 lysozyme. The OneG-Vali is implemented using CGI-Perl and it can be freely accessed and instantly used at http://sblab.sastra.edu/oneg-vali.html.
Introduction
Understanding structural interactions and energetics of fully folded proteins and partially unfolded forms (PUFs) existing in the unfolding and folding pathways of proteins are essential to specify the forces governing structural architectures, stability and biological functions of proteins under normal and pathological conditions as well.1–3 While fully folded proteins could be characterized at high resolution using various structural tools, structural characterizations of PUFs are being challenging as they need to be trapped by energetic barriers and also to be accumulated in amenable quantity in the study state and as well in kinetic processes.4–6 Moreover, most biophysical techniques and biochemical methods that characterize either fully folded or fully unfolded ensembles of proteins fail to detect PUFs as signals from the PUFs are swamped by that of folded or unfolded forms of proteins in those experimental conditions.7–9 In these contexts, especially cryptic intermediates (CIs), short-lived interceders populating sparsely between the folded and unfolded states of proteins, elude analyses of most traditional folding experiments.7,10 Proteins fold and unfold through distinct CIs in a sequential manner even under conditions favoring native state(s).8,11 Interestingly, kinetic and thermodynamic intermediates detected for several proteins in folding experiments resemble CIs characterized by NS H/D exchange methods in unfolding kinetics of the proteins under native conditions.11–14 Thus, structural characterizations and quantitative estimations of CIs are obviously essential to address structure–function relationships of proteins, to solve ‘Levinthal paradox’ and also to gain clues to folding pathways of proteins that adopt misfolded conformations.10,15–17
Fortunately, the CIs accumulating in the unfolding kinetics of proteins under native conditions can be qualitatively identified, structurally probed at residue level and quantitatively estimated by using NS H/D exchange methods in conjunction with multi-dimensional NMR and mass spectrometry techniques.18–22 To date, residue-specific free energies of exchange for more than 80 proteins have been studied using H/D exchange methods (in the absence of denaturants) and energetic landscapes of 16 proteins under their native conditions (in the presence of low denaturant concentration) have been characterized using NS H/D exchange methods.23 Notwithstanding the uniqueness of the methods (only available experimental strategies to date) on characterizing the CIs of proteins under native conditions, the methods are time consuming (days to several months) and require sound theoretical and experimental knowledge on the protein dynamics and H/D exchange mechanisms.8 Apart these factors, the methods are applicable only to proteins that are retaining their native fold throughout the course of experiments and furthermore the methods can only delineate unfolding pathways of proteins that are depicting distinct isotherm for each CI accumulating in the unfolding kinetics of the proteins.8,24 In these contexts, computational approaches will be excellent alternatives to the H/D methods.24,25 There are several programs to predict intrinsic/extrinsic exchange rates of backbone amide protons (NHs) of proteins and to estimate overall folding rates of polypeptides.25–31 To our best knowledge, OneG, a computational tool developed by the authors, is the only tool available to qualitatively predict cryptic intermediates/metastable states of proteins to date.32,33 In the present study, we demonstrate herein a novel computational tool, OneG-Vali, which determines the possible existence of cryptic intermediates (CIs) accumulating under native conditions of proteins in qualitative and as well quantitative manner. The tool also validates its prediction strength through combined analysis of multistate unfolding phenomenon predicted by the tool and pseudo two-state unfolding characterized by macroscopic methods for proteins. The tool is simple to use and is efficient to complete a successful run in fully automated manner using 4 prerequisite inputs (pdb files, ΔGHx, ΔGU and Cm of proteins – refer to Methods section). Furthermore, unique applications of the OneG-Vali on accounting the effect of cis–trans proline isomerization on estimating population of CIs defined by the tool/experimental methods have also been discussed in detail.
Methods
Defining foldons, global unfolding units and CIs of proteins
The OneG-Vali web-server has been designed using CGI-Perl.34 The function of the tool on predicting foldons of proteins is similar to the strategies described for OneG computational tool32,33 (ESI Fig. S1†). The OneG program is prerequisite of 4 inputs: atomic coordinates of proteins, ΔGHx (residue-specific free energy of exchange determined by using H/D exchange method in the absence of denaturant), ΔGU (free energy of unfolding) and ΔG*U (recalculated ΔGU upon appropriately treating the baselines of melting curves). In outline, the OneG program first detects all amide protons (NHs) that are hydrogen bonded in regular secondary structural elements of proteins on the basis of H-bond distance, H-bond angle, H-bond patterns and H-bond protections. Second, the program generates all possible residue pairs for the NHs and calculates distance in angstrom between the backbone nitrogen atoms of the two residues in each pair. The program then generates a ‘contact order matrix’ in which each pair is assigned either with the value of 1 or 0: the value of 1 is given to a pair when the distance between the two residues is within 7 Å otherwise 0 is given. Third, the program clusters the residue-pairs such that any pair in a group must have at least another pair having a residue common to each other. The program avoids redundancy in clustering the residue-pairs and generates atomic coordinate files in PDB format for residues in each cluster. If OneG finds more than a cluster for a protein, each cluster is distinct from other clusters in terms of structural contexts and consequently, each cluster is attributed to possible existence of a foldon in the unfolding kinetics of the protein.32,33 In these contexts, the OneG and OneG-Vali are differing from each other in the following two aspects: (i) the OneG-Vali considers all backbone amide protons (NHs) that are hydrogen bonded in regular secondary structures and as well possessing log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P (protection factor) of ≥2.0,35 whereas OneG considers NHs fulfilling the former criterion only; the later criterion used in the OneG-Vali helps only to increase prediction stringency on differentiating NHs that are hydrogen bonded in the regular secondary structures of proteins from NHs that establish hydrogen bonding on protein surfaces. (ii) OneG predicts cryptic intermediates in qualitative manner only whereas OneG-Vali predicts CIs in qualitative as well as quantitative manner. In addition to 4 inputs mentioned to the OneG, the OneG-Vali requires one more experimental parameter, ‘Cm’ (denaturant concentration wherein ΔGU is zero). Significances of each input to the programs are as follows: atomic coordinates of proteins are essential to identify NHs present in the regular secondary structures of proteins; ΔGHx values are essential to segregate NHs on the basis of H-bond protection, to map-out free energy coverage of each CI, to calculate free energy exchange (ΔGI) for each CI and also to determine order of CIs in the unfolding kinetics of proteins; Cm is essential to calculate population of each CI; ΔGU is essential to understand free energy discrepancies (ΔGHx vs. ΔGU) of proteins and as well to validate the predicted unfolding kinetics of proteins by the computational tools; the ΔG*U is an optional input for both OneG and OneG-Vali tools.
P (protection factor) of ≥2.0,35 whereas OneG considers NHs fulfilling the former criterion only; the later criterion used in the OneG-Vali helps only to increase prediction stringency on differentiating NHs that are hydrogen bonded in the regular secondary structures of proteins from NHs that establish hydrogen bonding on protein surfaces. (ii) OneG predicts cryptic intermediates in qualitative manner only whereas OneG-Vali predicts CIs in qualitative as well as quantitative manner. In addition to 4 inputs mentioned to the OneG, the OneG-Vali requires one more experimental parameter, ‘Cm’ (denaturant concentration wherein ΔGU is zero). Significances of each input to the programs are as follows: atomic coordinates of proteins are essential to identify NHs present in the regular secondary structures of proteins; ΔGHx values are essential to segregate NHs on the basis of H-bond protection, to map-out free energy coverage of each CI, to calculate free energy exchange (ΔGI) for each CI and also to determine order of CIs in the unfolding kinetics of proteins; Cm is essential to calculate population of each CI; ΔGU is essential to understand free energy discrepancies (ΔGHx vs. ΔGU) of proteins and as well to validate the predicted unfolding kinetics of proteins by the computational tools; the ΔG*U is an optional input for both OneG and OneG-Vali tools.
Qualitative and quantitative analyses of cryptic intermediates in the unfolding of proteins such as cytochrome c (PDB ID:1HRC; pH 7.0; 303 K), apocytochrome b562, (PDB ID:1APC; pH 4.5; 298 K), third domain of PDZ (PDB ID:1BE9; pH 6.3; 298 K) and T4 lysozyme (PDB ID:1L63; pH 5.6; 298 K) were examined by means of the OneG-Vali at conditions (pH and temperature) matching the NS H/D exchange experiments carried out to the proteins.36–40 At first, the OneG-Vali predicts various foldon units of proteins on the basis of their atomic co-ordinates by using ‘contact order matrix’ strategies32,33 and essential steps of the program are enumerated in Fig. 1. When the program identifies ‘n’ foldon units for a protein, the program defines ‘n − 1’ individual cooperative CIs to the protein, by default. Foldon unit that is composed of most slowly exchanging NHs is represented as globally exchanging/unfolding structural unit (GUU) and other foldon units are considered to represent various CIs of proteins. The CIs have been ordered on the basis of their free energies in descending manner. Free energy coverage for each CI is defined by taking into consideration of all NHs constituting respective CIs and free energy of exchange for each intermediate (ΔGI) is then averaged out to two largest ΔGHx of the residues in the respective intermediate. On the other hand, foldon units having similar free energy of exchange (within 0.4 kcal mol−1 difference) but distantly separated (>7 Å) are integrated and treated as single cooperative CI. Free energy exchange of the cooperative unit is calculated by averaging out ΔGI values of each CI in the integrated unit. For the purpose of clarity and forthright discussions, CIs are represented by residues defining respective isotherms of the CIs throughout the text and tables of the article, unless stated otherwise. It should also be mentioned that secondary structure of a CI accumulating in unfolding pathway of protein is structure of the protein with complete loss of native secondary structures of all foldons having free energy of exchange that are equal and as well less than that of the CI. Throughout the calculations, ΔGU, ΔGHx, ΔGI (free energies in kcal mol−1) and m-values (kcal mol−1 M−1, cooperative constants defined below) are considered in two decimal resolutions and Cm and MD (defined below) are represented in molarities with single decimal resolution.
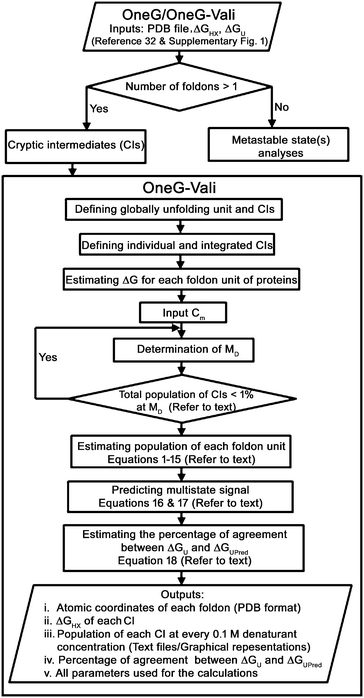 |
| | Fig. 1 Workflow diagram of OneG-Vali. Flowchart outlines key-steps of the OneG-Vali used to detect, estimate and validate cryptic intermediates of proteins under native conditions. | |
Quantitative estimations of CIs, folded and unfolded states of proteins
Unfolding cooperative constants for globally unfolding unit (mx) and each CI (mi) are calculated as shown in the following eqn (1) and (2), respectively (refer to ‘Results and discussion’ for the theoretical basis of the equations).wherein, ΔGHx is free energy exchange of global unfolding unit; ΔGmd is the free energy of exchange at MD and the value is calculated using the following relationship.| | |
ΔGmd = ΔGHx − (mx × MD)
| (3) |
wherein, MD is the concentration of denaturant at which total population of CIs is 1% or little greater than 1% (the concentration wherein melting transitions of proteins just begin) as the program calculates population of CIs at every 0.1 M denaturant concentrations. Equilibrium unfolding constant for global unfolding unit (KHx) and each CI (KI) are calculated using eqn (4) and (5), respectively.| | |
KHx = exp((mx × [D] − ΔGHx)/RT)
| (4) |
| | |
KI = exp((mi × [D] − ΔGI)/RT)
| (5) |
wherein, ‘R’ is gas constant and ‘T’ is absolute temperature in Kelvin. The program calculates population of CIs, folded and unfolded states of proteins at every 0.1 M denaturant concentration by using sequential unfolding model.10,11 For instance, according to this model, population of each CI is calculated for a protein having 4 foldon units (one GUU and three CIs) as shown below, herein.| |
 | (6) |
| | |
[CI3] = K3/(1 + K3 + K2 + K1 + KHx)
| (7) |
| | |
[CI2] = K2/(1 + K3 + K2 + K1 + KHx)
| (8) |
| | |
[CI1] = K1/(1 + K3 + K2 + K1 + KHx)
| (9) |
| | |
[N] = 1/(1 + K3 + K2 + K1 + KHx)
| (10) |
| | |
[U] = KHx/(1 + K3 + K2 + K1 + KHx)
| (11) |
wherein, equilibrium constants K3, K2, K1 and KHx are calculated as shown below.| | |
K2 = [CI2]/[N] = K3 × K23
| (13) |
| | |
K1 = [CI1]/[N] = K3 × K23 × K12 = K2 × K12
| (14) |
| | |
KHx = [U]/[N] = K3 × K23 × K12 × KU1 = K1 × KU1
| (15) |
Predicting multistate unfolding curves of proteins
As the NS H/D experiments reveal secondary structural contacts of CIs in high resolution, population of CIs, folded and unfolded states of proteins predicted by the tool with respect to denaturant concentrations could be used to predict multistate melting curves that would be measured by various optical techniques monitoring the equilibrium unfolding of proteins through average measurements of changes in the secondary structural contents of the proteins.9 The normalized multistate unfolding of protein was predicted according to following equations:| |
 | (16) |
wherein, SD is the normalized signal for the multistate-mixture of proteins at ‘D’ concentration of denaturant; N and CI denote native state (folded conformation) and cryptic intermediate; WN and Wn are weighted factors (‘i’ and ‘n’ denote total number and position of CI – ranked on the basis of their free energy of exchange from highest to lowest energies – respectively). While value of WN is 1 for folded state, values of Wn for CIs can be calculated as shown in the following equation:| |
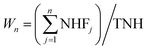 | (17) |
wherein, NHFj and TNH represent total number of NHs present in the ‘jth’ foldon unit and total number of NHs considered for the prediction of CIs in proteins by the OneG-Vali, respectively. The OneG-Vali determines free energy of unfolding (ΔGUPred) for the predicted multistate melting curve (as if pseudo two-state melting curve) by multiplying values of mPred (unfolding cooperative constant, which is slope of plot depicting ΔGUPred at 20% and 80% of native population vs. respective denaturant concentrations (ΔD), i.e. mPred = ΔΔGUPred/ΔD) and CmPred (denaturant concentration at which population of fully folded state is 50%). Percentage of agreement between the multistate melting curve calculated by the tool and pseudo two-state melting curve determined by using optical probes for proteins is assessed by using eqn (18).| | |
Percentage of agreement = [(ΔGHx − ΔGUPred)/(ΔGHx − ΔGU)] × 100
| (18) |
When all 4 prerequisite parameters are given as inputs, the tool completes a successful run within a few minutes for a protein having 100 amino acids and generates individual structural coordinates for each foldon unit of proteins in ‘pdb’ format. Moreover, population of each CI in ‘txt format’ (at every 0.1 M denaturant concentration covering a default range from 0 to 7.0 M), multistate melting curves (in graphical and as well in txt formats), percentage of agreement between ΔGU & ΔGUPred and all calculated parameters (ΔGI, mi of each CI, ΔGHx, mx, MD, ΔGmd, Wn, ΔGUPred and mPred) can also be retrieved from the OneG-Vali for every successful run. The program can be easily accessed and instantly used without prior registration or permission from the authors. Off-line version of the OneG-Vali can also be freely obtained from the authors upon request and the tool can be successfully installed and executed in systems with minimum preferable configuration of 2 GB RAM, 250 GB HD and a dual core processor.
Results and discussion
Rationalizations on the structural contexts of cryptic intermediates determined by experimental and computational methods
We have recently shown that CIs/metastable states accumulating in the unfolding kinetics of proteins under native conditions could be well predicted by means of the OneG computational tool.32,33 The OneG-Vali defines possible existence of CIs in the unfolding kinetics of proteins and as well estimates population of the CIs with respect to denaturant concentration. Of the 16 proteins characterized using NS H/D exchange methods to date, proteins such as cytochrome c,7,36 apocytochrome b562,37 third domain of PDZ38 and T4 lysozyme39,40 possessed adequate inputs for the OneG-Vali. Table 1 depicts structural contexts and stability of various foldons for the 4 proteins obtained from the experimental and computational methods. Graphical comparisons of various foldons of the 4 proteins detected by NS H/D exchange method and predicted by the OneG-Vali computational tool are also illustrated using their respective three-dimensional structures (Fig. S2–S5†). From a quick inspection to the Table 1, it is obvious that predictive success of the tool is quite impressive and overall structural contexts of various foldons defined from the H/D labeling methods are matching well with OneG-Vali predictions of the proteins. The agreement between the CIs predicted by the OneG tool and CIs detected by using NS H/D exchange methods for cytochrome c and apocytochrome b562 have been already documented in the literature.32 But, there were modest differences between the two methods (OneG-Vali & NS H/D exchange) on defining CIs for third domain of PDZ (PDZ) and the dispute are mainly due to differences in minimum-residue cutoffs used to define CIs by the methods. The N- & C-termini of the PDZ could be experimentally defined as a CI of the protein by having only one representative residue (Lys 97) for the whole region.38 Though the OneG-Vali identified the residue as a probe, the region was not declared as a CI of the protein because the program requires minimum 3 residues to define a foldon and this condition is based on the fact that at least 3 residues are needed to form either a stable helical turn or sheet conformations in proteins.41 Moreover, the OneG-Vali accounts more probes (NHs) to define distinct foldons of proteins than the number of probes identified from the NS H/D exchange experiments. Because, all NHs of proteins that are acting as probes for H/D exchange in the absence of denaturant may not act as feasible probes for H/D exchange in the presence of various denaturant concentrations owing to denaturant perturbations on structural contacts of the proteins.42–44 The rationalization also holds good to address some modest differences on defining structural boundaries of CIs in the T4 lysozyme by the manual and computational methods. To this extent, the strength of the OneG-Vali is reliable on qualitatively identifying the possible existence of CIs of the proteins. Furthermore, quantitative estimations of the CIs and their validations as we have demonstrated in the following sections for the four proteins strongly justify the veraciousness of the OneG-Vali on predicting unfolding kinetics of the proteins under native conditions.
Table 1 Quantitative estimations of cryptic intermediates in proteins by using the NS H/D exchange methods and OneG-Vali computational tool
| Proteins |
NS H/D Exchange results |
OneG-Vali results |
| FUa |
Residues & regionsb |
ΔGc |
CIPd |
TPe |
FUa |
Residues & regionsb |
ΔGc |
CIPd |
TPe |
| Foldon Units. Residue numbers and their respective structural contexts are given; GUU and CI denote globally unfolding unit and cryptic intermediate, respectively; helices and strands are denoted by ‘H’ and ‘S’ respectively. Free energy exchange values of foldon units in kcal mol−1. Maximum population of each CI. Total population of CIs. Not determined by experimental methods. |
| Cytochrome c |
4 |
GUU: 7 8 10–13 19 91–101 [N- and C-terminal] |
12.95 |
|
16 |
3 |
GUU: 7 9–15 18 91–99 101 102 [N- and C-terminal] |
12.95 |
|
16 |
| CI1: 32 33 65–70 [60's helix] |
10 |
7 |
|
|
CI1: 64 65 67–70 73–75 85 [60's helix and 70's loop] |
10.39 |
15 |
|
| CI2: 36 37 59 [region spanning 36–61] |
7.4 |
7 |
|
|
CI2: 52–54 [region spanning 36–61] |
4.98 |
1 |
|
| CI3: 74 75 85 [70's loop] |
6 |
2 |
|
|
|
|
|
|
| Apocytochrome b562 |
3 |
GUU: 32–37 70 71 75–77 [H2 & H3] |
NDf |
|
|
3 |
GUU: 11 13 14 16 17 26–30 32–43 [H1 & H2] |
5.54 |
|
18 |
| CI1: 87–91 [H4] |
|
|
|
|
CI1: 68–72 75 76 79–81 [H3] |
4.95 |
14 |
|
| CI2: 14–17 [H1] |
|
|
|
|
CI2: 87–91 93 [H4] |
4.40 |
4 |
|
| Third domain of PDZ |
2 |
GUU: 30 42 65 83 90 [S1–S5 & H2] |
ND |
|
|
2 |
GUU: 16 18 20 32 41–43 60–64 66 69 71 92–95 [S1–S5] |
8.81 |
|
10 |
| CI1: 97 [N- & C-terminal] |
|
|
|
|
CI1: 79–84 [H2] |
6.69 |
10 |
|
| T4 lysozyme |
2 |
GUU: 5 77 98 106 153 [H1 of N- & C-terminal helix] |
ND |
|
|
4 |
GUU: 62–81 85 87–91 96–107 111 112 121–125 129–135 140–142 146–156 [C-terminal] |
18.0 |
|
14 |
| CI1: 16 27 45 50 [N- terminal H2 and sheet] |
|
|
|
|
CI1: 5–12 14 [H1] |
14.2 |
12 |
|
| |
|
|
|
|
CI2: 18 20 27 31 33 [N-terminal sheet] |
7.30 |
1 |
|
| |
|
|
|
|
CI3: 42–51 [N-terminal H2] |
6.75 |
1 |
|
Computational strategies for estimating population of cryptic intermediates
Two strategies have been conceptualized to estimate population of CIs using the OneG-Vali: (i) Cm value (mid-point of melting transition) of pseudo two-state equilibrium unfolding of a protein studied by optical techniques and Cm values of residues exchanging by global unfolding events of the protein are deemed to be same (ii) H/D exchange isotherms for various foldon units of proteins are considered to converge at a denaturant concentration (MD – meeting point of isotherms), wherein total population of intermediates is 1% or little higher than 1% (refer to Methods section). In other words, proteins just begin to melt at the MD and the CIs, folded and unfolded conformations are reasoned to be around 1%, 98% and 1% respectively at the denaturant concentration. These two strategies could be well rationalized as demonstrated herein. The former strategy is on the fact that CIs accumulating in the unfolding transition regions of equilibrium experiments exclusively affect free energy (m × Cm) of protein through ‘m’ value (cooperative constant) and their effect on Cm is negligible.9,45 Interestingly, this is evidently proven through NS H/D exchange studies carried out on oxidized equine cytochrome c, the only protein for which population of cryptic intermediates have been experimentally estimated to date.9 Free energy of unfolding (ΔGU determined by optical methods) and free energy of exchange (ΔGHx determined from NS H/D exchange methods) of the protein were reported as 10 kcal mol−1 and 12.8 kcal mol−1, respectively and cooperative constants (m values) determined by the optical and H/D exchange methods were reported as 3.6 kcal mol−1 M−1 and 4.6 kcal mol−1 M−1, respectively suggesting the Cm values determined by both methods were exactly same (2.78 M) for global unfolding of the protein. The discrepancy between the ΔGU and ΔGHx has been attributed to the existence of three CIs in the unfolding kinetic of the protein as examined by NS H/D exchange methods in conjunction with NMR techniques.9,45
The later strategy is based on the fact that CIs and unfolded conformations of proteins, which infinitesimally exist under native conditions, are significantly accumulating in the melting transitions.9,11 Population of cryptic intermediates gradually increases in melting transitions and reaches maximum at near Cm concentrations and beyond the concentrations, CIs gradually degrade to unfolded states with respect to denaturant concentrations.8,9 In NS H/D exchange experiments, each CI can be represented by a unique isotherm, so that energy-well for each CI can be defined in the energy landscape of the protein under native condition. However, kinetic barriers of CIs cannot be determined by the method itself and the kinetic barriers do not affect detections of the CIs by H/D exchange under native conditions as shown for several proteins in the literature.8,10 Model energy diagrams for the unfolding kinetics of a protein in which 2 CIs exist under native conditions are schematically shown in Fig. 2, which exemplifies accumulations and degradations of the CIs before and after Cm concentration, respectively. The free energy difference between the CIs and folded form of proteins will gradually decrease with increasing concentration of denaturants within a range wherein added denaturant does not alter the population of CIs, folded and unfolded states of proteins. At these low concentrations of denaturants, the CIs occupy energy levels that are lower than the energy level of the unfolded form (UF), as the CIs are more stable than the UF under the conditions (Fig. 2). In the unfolding transition regions, UF populates in larger quantity than the population of CIs as the conditions favor denaturation of proteins. Because of this phenomenon, the energy well of UF approaches the folded form at Cm concentration, before CIs approaches it, and in turn the CIs are not being detectable in the traditional equilibrium folding experiments.8,9 Beyond the Cm concentration, CIs approaches the energy level of folded form one by one as per the order of their stabilities and at high concentration of denaturant, the folded and UF overtake population of the CIs and occupy energy landscape predominantly (Fig. 2).
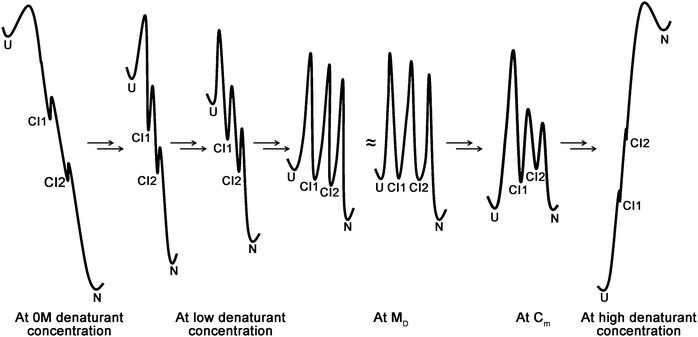 |
| | Fig. 2 Model energy diagrams for denaturant-induced protein unfolding. Schematic energy diagrams for folded (N), unfolded (U) and two cryptic intermediates (CIs) states of a protein under equilibrium unfolding conditions at various denaturant concentration. The energy wells and kinetic barriers are arbitrary and intended to illustrate the relationships among the various states at a particular condition only. The depth of energy wells and magnitude of kinetic barriers of a particular state at various conditions do not necessarily bring any series relationships in the figurative representations. | |
In the present study, the MD is defined as a concentration of denaturant (used in the equilibrium unfolding studies of proteins), wherein the total population of CIs is about 1% (refer to Methods section). In other words, MD is a denaturant concentration, where the unfolding transitions of proteins just begin and each CI of proteins begins to accumulate at MD by its unique energy barrier. The energy levels of all CIs are lower than that of UF at just before MD and higher than that of UF at Cm. As the UF and CIs of proteins just begin to accumulate in comparable quantity to each other at MD, the energy wells of UF and CIs should be same or closely similar to each other with comparable energy barriers (Fig. 2). When the MD was set at around 1% intermediates accumulation, population of CIs predicted by the OneG-Vali was less than 20% for all proteins considered in the present study (Table 1). Moreover, total population of experimentally detected CIs of cytochrome c was 16% (calculated by using experimental parameters). Strikingly, total population of CIs predicted by the OneG-Vali for the protein was also found to be 16%. Furthermore, isotherms of 4 foldon units of the protein detected experimentally are converging closely around 2.15 M GdnDCl,9 which is in good agreement with the calculated MD value of 2.20 M for the protein by the tool. Interestingly, when MD (at around 1% of total population of CIs) was increased to MD + 0.2 M, the apocytochrome b562, T4 lysozyme, cytochrome c and third domain PDZ showed maximum CIs population of 36, 29, 26 and 15% respectively. Similarly, when MD was set at around 2% of total population of CIs, the apocytochrome b562, T4 lysozyme, cytochrome c and third domain PDZ showed maximum CIs population of 27, 20, 21 and 15% respectively. The CIs population of all the 4 proteins were found to increase with increasing MD values and the data become of little meaning, because, drastic accumulation of CIs (>20%) is unlikely under equilibrium unfolding experiments. Indeed, all the proteins need not necessarily accumulate CIs of 20% under native conditions. Moreover, isoenergetic wells of CIs and unfolded state with similar energy barriers are also very unlikely to shift toward Cm, since unfolding conditions are favored at high denaturant concentration (Fig. 2). On the other hand, the MD can be fine-tuned around CIs population of about 1% (by gradually increasing the MD value at every 0.1 M; the program offers the feature to perform the calculations as an option) and the estimated population of CIs of proteins can be thoroughly validated by combined comprehensive analysis of the proteins multistate unfolding predicted by the program and equilibrium unfolding of respective proteins estimated from optical methods. Strikingly, the analyses carried out on the four proteins considered in the present study (refer to next section) apparently vindicated that the rationalization made on determining the MD (concentration of denaturant wherein total population of CIs is about 1%) is reliable to quantify CIs of the proteins.
Comparative analysis of multistate and pseudo two-state protein unfolding
Various structural stability parameters obtained from the OneG-Vali analyses, equilibrium unfolding and NS H/D exchange studies of proteins such as cytochrome c, apocytochrome b562, PDZ and T4 lysozyme are depicted in Table 2. Using the structural features and population of foldons of the proteins predicted by the tool, multistate unfolding melting curves for the respective proteins were calculated and free energy of unfolding estimated from the multistate curves is represented as ΔGUPred (refer to Methods section). Denaturant-induced pseudo two-state unfolding curves determined by optical probes and multistate melting curves predicted by the OneG-Vali for the 4 proteins considered in the present study are depicted in Fig. 3. Direct comparisons of the ΔGU and ΔGUPred of the proteins are in good agreement ((ΔGUPred/ΔGU) × 100 = 108 ± 10, Table 2) suggesting that prediction efficiency of the tool on mapping out the unfolding pathways of the proteins is quite impressive. Moreover, percentage of agreement between the ΔGU (free energy of unfolding estimated by optical methods) and ΔGUPred (free energy of unfolding calculated by the OneG-Vali) for each protein were also stringently determined by using eqn (18), which explicitly reveals the extent to which the ΔGUPred of a protein addresses the apparent discrepancy between the ΔGU and ΔGHx (free energy of exchange determined from NS H/D exchange methods) of the protein. Interestingly, the free energy discrepancies (between ΔGU and ΔGHx) of cytochrome c, PDZ and T4 lysozyme could be well accounted (to extent of 96%, 94% and 82%, respectively) by the respective ΔGUPred of the proteins (Table 2). However, ΔGUPred of apocytochrome b562 could address only 67% of free energy discrepancy for the protein implying that total population of CIs of the proteins are presumably underestimated by the computational tool. The free energy discrepancy of the protein may be well addressed by taking into account the effect of cis–trans proline isomerization in calculating multistate melting curve of the protein, as the OneG-Vali determines the ΔGUPred for protein without accounting the effect of proline isomerization by default.
Table 2 Validations of cryptic intermediates of proteins predicted by the OneG-Vali
| Protein |
NS H/D exchange resultsj |
OneG-Vali resultsj |
| ΔGHx |
ΔGU |
mua |
NCb |
TPc |
ΔGd |
me |
%f |
Without proline correction |
With proline correction |
| NCb |
TPc |
ΔGd |
me |
%f |
TPc |
ΔGd |
me |
%f |
| ‘mu’ represents cooperative constant determined from the experimental method. Number of CIs. Total population of CIs. ‘ΔG’ represents free energy of unfolding predicted by OneG-Vali for multistate melting signal (ΔGUPred). ‘m’ represents cooperative constant predicted by OneG-Vali for multistate melting signal (mPred). Percentage of agreement between ΔGUPred and ΔGU. Percentage of agreement is calculated by using experimental data of the protein. Not determined. Not applicable. Free energy of unfolding and ‘m’ values are given in kcal mol−1 and kcal mol−1 M−1, respectively. |
| Cytochrome c |
12.95 |
10.0 |
3.57 |
3 |
16 |
10.14 |
3.74 |
95g |
2 |
16 |
10.12 |
3.68 |
96 |
14 |
10.27 |
3.72 |
91 |
| Apocytochrome b562 |
5.54 |
3.3 |
3.0 |
2 |
NDh |
|
|
NAi |
2 |
18 |
4.03 |
3.84 |
67 |
23 |
3.62 |
3.48 |
86 |
| Third domain of PDZ |
8.81 |
7.4 |
2.47 |
1 |
ND |
|
|
NA |
1 |
10 |
7.49 |
2.54 |
94 |
12 |
7.05 |
2.39 |
125 |
| T4 lysozyme |
18.0 |
13.5 |
4.82 |
1 |
ND |
|
|
NA |
3 |
14 |
14.32 |
5.17 |
82 |
15 |
14.13 |
5.10 |
86 |
 |
| | Fig. 3 Protein unfolding examined by optical probes and OneG-Vali. Denaturant-induced melting curves (solid lines) determined by optical probes and multistate unfolding curves (dashed lines, uncorrected to effect of proline isomerization) predicted by the OneG-Vali computational tool are depicted for cytochrome c, apocytochrome b562, third domain of PDZ and T4 lysozyme. Total population of cryptic intermediate(s) (uncorrected to proline isomerization) accumulating in the unfolding kinetics of the proteins with respect to denaturant concentration as predicted by the computational tool is shown in filled circles. | |
Amide bonds of standard amino acids in polypeptide chains are exclusively in trans conformation in folded states, whereas imide bond of proline (Xaa–proline, wherein Xaa stands for any standard amino acid) adopts cis or trans conformations much more equally in the folded forms of proteins.46,47 Similarly, amide bonds prefer negligible percentage of (about 0.03%) cis-conformations in the unfolded states, whereas imide bond (Xaa–Pro) prefers remarkable percentage of cis-conformations in the unfolded states and the percentage varies (6–38%) depending on the chemical properties of the residue preceding proline in the imide bond.46 Hence, the folded forms and as well CIs consisting of proline residues may undergo cis–trans proline isomerization in the unfolding kinetics of proteins monitored by NS H/D methods. Since the cis–trans proline isomerization is a slow process comparing the exchange rates of NHs in proteins, the effect of the proline isomerization cannot be detected by the NS H/D exchange methods.47 The effect of cis–trans proline isomerization on the ΔGHx of folded and CIs states of proteins can be readily calculated by using well-established reports.9,47 In order to account the effect of cis–trans proline isomerization, the program offers options to recalculate the multistate unfolding curves of proteins provided Xaa–proline imide bond(s) of each CI of the proteins are defined by the user. Interestingly, after accounting the effect of proline isomerization, the ΔGUPred of apocytochrome b562 was found to be 3.6 kcal mol−1 and it addressed 86% of the free energy discrepancy of the protein. Apocytochrome b562 is a four helix bundle protein and consists of four trans imide bonds with prolines located at positions 45, 46, 53 and 56 (Thr44–Pro45, Pro45–Pro46, Ser52–Pro53 and Ser55–Pro56). The program predicted a global unfolding unit (GUU) and two CIs (in total three foldons) of the protein: the GUU consisted of residues from helix I & II; CI1 consisted of residues from helix III; CI2 consisted of residues from helix IV (refer to ‘Methods’ and Table 1). The ΔGHx values of the GUU, CI1 and CI2 were 5.54, 4.95 and 4.40 kcal mol−1, respectively, without accounting the effect of proline isomerization and the total population of CIs was found to be 18% (Tables 1 and 2). The CI1 contains two trans prolines (Ser52–Pro53 and Ser55–Pro56) and CI2 was bereft of proline residues. After accounting the effect of prolines, the recalculated ΔGHx values of the GUU, CI1 and CI2 were 5.32, 4.82 and 4.40 kcal mol−1, respectively and the total population of CIs was also recalculated to be 23% (CI1 = 17% and CI2 = 6%). This clearly indicated that significance of the proline isomerization on estimating the population of CIs and as well addressing the discrepancy between the ΔGHx and ΔGU of proteins, in general.
The OneG-Vali predicted four foldon units of T4 lysozyme: GUU, CI1, CI2 and CI3 of the protein were constituted by residues from C-terminal helix, helix1, N-terminal sheet and helix2, respectively (Table 1). The protein has three Xaa–Pro peptide bonds (Ser36–Pro37, Lys85–Pro86 and Thr142–Pro143) in trans conformations. Of the three prolines, no proline was belonging to the CI1 of the protein and Pro37 is in close proximities to residues of the CI2 and CI3 as well. Table 2 provides details on the ΔGHx and population of the CIs that are uncorrected and as well corrected to the effect of proline isomerization for the protein. Upon accounting the effect, the ΔGHx of the GUU, CI1, CI2 and CI3 were 17.84, 14.14, 7.24 and 6.69 kcal mol−1, respectively and total population of the CIs was estimated to be 15%, which is just 1% higher than that of the CIs calculated without considering the proline roles on unfolding process of the protein (Table 2). Values of ΔGUPred corrected (14.1 kcal mol−1) and uncorrected (14.3 kcal mol−1) to proline isomerization could account 86% and 82%, respectively to the free energy discrepancy (ΔGHx vs. ΔGU) of the protein suggesting proline impacts were modest on the protein unfolding kinetics. Similarly, the OneG-Vali predicted three foldons of cytochrome c as follows: GUU consisted of residues from the N- and C-termini helices; CI1 consisted of residues from 60's helix and 70's loop regions; CI2 consisted of residues from region spanning 36–61 (Table 1). The protein has 4 prolines located at 30, 44, 71 and 76 positions and all are in trans conformation. Prolines at 71 and 76 were situated in CI1 and no proline residue was present in the CI2 of the protein. Values of ΔGHx corrected to proline isomerization were 12.66, 10.24 and 4.98 kcal mol−1 for GUU, CI1 and CI2 of the protein, respectively. Using the proline corrected ΔGHx values, the ΔGUPred of the protein was estimated as 10.3 kcal mol−1, which accounted 91% of the discrepancy between ΔGHx and ΔGU of the protein (Table 2). As discussed above, the discrepancy could be addressed to the level of 96% by the ΔGUPred calculated without taking into consideration of effect of proline residues suggesting that proline residues of the protein did not cause remarkable effect on estimating multistate unfolding from the CIs of the protein. However, the proline residues of PDZ showed prominent impacts on estimating free energy of unfolding of the protein. Accounting the effect of proline isomerization to the unfolding of PDZ brought results that are different from the observations discussed above for apocytochrome b562, cytochrome c and T4 lysozyme. Both NS H/D exchange and OneG-Vali methods suggested existence of single CI in the unfolding kinetics of PDZ under native conditions. The CI and GUU of the PDZ were consisting of residues from helix2 and strands 1–5 of the protein, respectively (Table 1). The protein has four trans Xaa–Pro peptide bonds (Ile7–Pro8, Glu10–Pro11, Gly45–Pro46 and Lys93–Pro94) and none of them belonging to structural contexts of CI1 of the PDZ. When the proline corrected ΔGHx (8.55 kcal mol−1) of the GUU was used to predict the protein unfolding, the estimated total population of CI1 and ΔGUPred of the protein were 12% and 7.1 kcal mol−1, respectively (Table 2). As per the eqn (18), the percentage of agreement between the ΔGU and ΔGUPrerd of the protein was 125% indicating that accumulation of the CI in the protein unfolding has been overestimated after accounting the effect of proline isomerization. These observations undoubtedly suggest that exact contributions of Xaa–Pro imide bonds on the equilibrium among the folded, CIs and unfolded states of proteins under native conditions are crucial to authentically address the discrepancies between the free energies determined by two different methods (especially for ΔG estimations from macroscopic and microscopic probes).
Multistate unfolding curves of apocytochrome b562 calculated with and without taking into consideration of proline isomerization were compared to the pseudo two-state unfolding of the protein determined by optical probes and the analyses revealed that the former one is in excellent agreement (86%) with the experimental data than the later one (67%) with the experimental data (Table 2 and Fig. 3). Interestingly, the effect of cis–trans proline isomerization did not cause any remarkable differences on predicting multistate unfolding curves of T4 lysozyme and cytochrome c (Table 2). In contrary, while the ΔGUPred calculated without accounting the proline isomerization for PDZ was in excellent agreement (94%) with the ΔGU (determined by denaturant-induced unfolding studies) of the protein, the ΔGUPred of the protein corrected to the effect of proline isomerization did not show good agreement (125%) with the ΔGU of the protein (Table 2). Hence, accounting cis–trans proline isomerization on predicting protein unfolding from data of H/D exchange experiments may significantly alter agreements between the ΔGU and ΔGUPred of proteins. In these contexts, it should be mentioned that the program accounts the proline effect in protein unfolding using Kpro values from studies of model compounds.46 Though the values of Kpro estimated from the model compounds account reasonably the cis–trans isomerization of Xaa–Pro peptide bonds in the unfolded states of most proteins,46,47 the KPro estimated based on the model compounds in a set of particular experimental conditions may not necessarily be a true representation to Xaa–Pro of proteins in a totally different solution conditions.48 To tackle this aspect, the program provides options to use KPro determined from the studies on specific proteins (inputs from users) for calculating the ΔGUPred of the proteins. It should be mentioned that overall percentage agreements between the ΔGU and ΔGUPred of the 4 proteins considered in the present study were found to be 97 ± 19 and 85 ± 13 (as calculated by eqn (18)) upon accounting and not accounting the effect of proline isomerization on predicting multistate unfolding curves for the 4 proteins, respectively. In these backgrounds, the strategies (‘contact order matrix’ and ‘MD – meeting point of isotherms’) used in the OneG-Vali to define foldons and as well to estimate population of CIs are highly reliable to probe the unfolding kinetics of proteins under native conditions. Interestingly, overall percentage agreements between the ΔGU and ΔGUPred of the 4 proteins considered in the present study were also found to be 135 ± 13 and 113 ± 17 (without accounting proline isomerization effect), when the MD was set at MD + 0.2 M and at 2% CIs accumulation, respectively and the overall agreements in both cases became very worse upon accounting proline isomerization in the calculations. Thus, it is also worthy to mention that MD defined either at higher (MD + D or ≈2% CIs accumulation) or at lower (MD − D) denaturant concentration (D) would significantly affect percentage agreement between ΔGU and ΔGPred of proteins, in general.
Besides predicting the unfolding pathways of proteins, the OneG-Vali also facilitates generating multistate melting curves to experimentally defined CIs of proteins provided free energy of unfolding, cooperative unfolding constant and weighted factor for each CI of the proteins are given as inputs. For instance, the tool predicted a multistate melting curve based on the experimental data reported for cytochrome c, the only protein for which population of cryptic intermediates have been experimentally estimated to date.7,9 NS H/D exchange studies on the protein uncovered existence of four foldons in the protein unfolding under native conditions: GUU consisted of residues from N- and C-termini helices; CI1 consisted residues from 60's helix; CI2 and CI3 consisted of residues from regions spanning from 36–61 and 70–85, respectively. The ΔGHx of the CI1, CI2 & CI3 were 10, 7.4 & 6.0 kcal mol−1, respectively; cooperative unfolding constants were 3.21, 2.29 and 1.50 kcal mol−1 M−1 for CI1, CI2 and CI3, respectively; weighted factors for the CI1, CI2 & CI3 were 0.61, 0.78 and 0.90, respectively. Using the experimental data, the OneG-Vali calculated population of CI1, CI2 & CI3 to be 7, 7 & 2% (16% in total), respectively and the ΔGUPred calculated from the multistate unfolding curve computed for the protein was found to be 10.14 kcal mol−1, which accounted 95% of the discrepancy between the ΔGHx and ΔGU of the protein (effect of cis–trans proline isomerization was not considered). These data were in excellent agreement with the OneG-Vali predictions for the protein. As discussed above, the program predicted two CIs of the protein, estimated 16% population of the CIs and validated that the CIs accounted 96% (ΔGUPred was 10.1 kcal mol−1) of discrepancy between the ΔGHx (13.0 kcal mol−1) and ΔGU (10.0 kcal mol−1) of the protein. The comparative analyses are also fortifying that the OneG-Vali is a reliable computational tool for quantifying CIs existing in native unfolding of proteins and as well on addressing the discrepancy between the ΔGU and ΔGHx of proteins.
Conclusions
To our best knowledge, the OneG-Vali is a unique computational tool (only available program to date) of this kind for qualitatively and quantitatively predicting CIs that may presumably accumulate in the unfolding kinetics of proteins under native conditions. When all 4 prerequisite parameters (atomic coordinates, ΔGHx, ΔGU, and Cm of proteins) are available, the tool completes a successful run within a few minutes and structural coordinates of foldons, population of each CI and all calculated parameters are directly downloadable in appropriate formats from the webserver. The tool can also be used to validate CIs characterized by NS H/D exchange methods. In addition, effect of cis–trans proline isomerization on estimating population of CIs (detected either by experimental or computational methods) can be calculated by using the OneG-Vali. Moreover, the tool can provide more probes representing CIs than that from H/D exchange studies, as structural prediction of CIs by the tool is mainly depending on the native 3D structures of proteins and the information in turn may presumably be useful to understand effect of denaturant on the structural dynamics relieving hydrogen bond protections for certain NHs of proteins under the NS H/D exchange experiments. Undoubtedly, the tool will be an excellent alternative to map out the energy landscapes of proteins that are not compatible for NS H/D exchange experiments owing to the experimental solution conditions causing association/aggregation/degradation of proteins.49 In case, misfolding of the proteins are principal factors of prion diseases,50–52 applications of the tool may play significant roles on addressing mechanisms of unfolding kinetics of the proteins53,54 through on identifying cryptic intermediates and estimating population of the CIs. Moreover, conflicts that may bob up between the predictions and experimental observations (especially in terms of total number of NHs) on the unfolding kinetics of proteins may pave a way to ameliorate various concepts for understanding the exchange phenomena of NHs55 in a precise manner. In these backgrounds, we do anticipate a great scope to extend the applications of the OneG-Vali on calculating folding dynamics of NHs in proteins in the near future.
Acknowledgements
We would like to express our gratitude to Prof. P. Thomas Muthiah, Department of Chemistry, Bharathidasan University, India and Prof. S.W. Englander, University of Pennsylvania, USA for generously sharing their views on the sequential/independent pathways of protein folding. This research work is supported by the research grant (09/1095/(0004)/2013/EMR-I) from the Council of Scientific & Industrial Research, New Delhi, India. We also sincerely thank the anonymous referees for constructive comments on an early version of the manuscript.
Notes and references
- R. L. Baldwin and G. D. Rose, Trends Biochem. Sci., 1999, 24, 26–33 CrossRef CAS.
- A. R. Dinner, A. Salib, L. J. Smitha, C. M. Dobson and M. Karplus, Trends Biochem. Sci., 2000, 25, 331–339 CrossRef CAS.
- T. K. S. Kumar and C. Yu, Acc. Chem. Res., 2004, 37, 929–936 CrossRef CAS PubMed.
- T. E. Creighton, Curr. Biol., 1991, 1, 8–10 CrossRef CAS.
- C. M. Dobson and P. A. Evans, Nature, 1988, 335, 666–667 CrossRef CAS PubMed.
- S. Gianni, Y. Ivarsson, P. Jemth, M. Brunori and C. Travaglini-Allocatelli, Biophys. Chem., 2007, 128, 105–113 CrossRef CAS PubMed.
- Y. Bai, T. R. Sosnick, L. Mayne and S. W. Englander, Science, 1995, 269, 192–197 CAS.
- Y. Bai and S. W. Englander, Proteins, 1996, 24, 145–151 CrossRef CAS.
- L. Mayne and S. W. Englander, Protein Sci., 2000, 9, 1873–1877 CrossRef CAS PubMed.
- Y. Bai, Biochem. Biophys. Res. Commun., 2003, 305, 785–788 Search PubMed.
- S. W. Englander, L. Mayne and J. N. Rumbley, Biophys. Chem., 2002, 101–102, 57–65 CrossRef CAS.
- Y. Bai, Chem. Rev., 2006, 106, 1757–1768 CrossRef CAS PubMed.
- A. K. Chamberlain and S. Marqusee, Structure, 1997, 5, 859–863 CrossRef CAS.
- A. R. Fersht, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 14121–14126 CrossRef CAS PubMed.
- R. L. Baldwin, Annu. Rev. Biophys., 2008, 37, 1–21 CrossRef CAS PubMed.
- U. Nath and J. B. Udgaonkar, Curr. Sci., 1997, 72, 180–191 CAS.
- K. W. Plaxco and C. M. Dobson, Curr. Opin. Struct. Biol., 1996, 6, 630–636 CrossRef CAS.
- T. M. Raschke and S. Marqusee, Curr. Opin. Struct. Biol., 1998, 9, 80–86 CAS.
- S. W. Englander, Science, 1993, 262, 848–849 CAS.
- S. W. Englander, T. R. Sosnick, J. J. Englander and L. Mayne, Curr. Opin. Struct. Biol., 1996, 6, 18–23 CrossRef CAS.
- D. M. Ferraro, N. Lazo and A. D. Robertson, Biochemistry, 2004, 43, 587–594 CrossRef CAS PubMed.
- G. S. Anand, C. A. Hughes, J. M. Jones, S. S. Taylor and E. A. Komives, J. Mol. Biol., 2002, 323, 377–386 CrossRef CAS.
- T. Richa and T. Sivaraman, Int. J. Res. Pharm. Sci., 2013, 4, 550–562 Search PubMed.
- S. W. Englander, L. Mayne and M. M. G. Krishna, Q. Rev. Biophys., 2007, 40, 287–326 CrossRef CAS PubMed.
- V. J. Hilser and E. Freire, J. Mol. Biol., 1996, 262, 756–772 CrossRef CAS PubMed.
- G. G. Tartaglia, A. Cavalli and M. Vendruscolo, Structure, 2007, 15, 139–143 CrossRef CAS PubMed.
- T. Richa and T. Sivaraman, J. Pharm. Sci. Res., 2012, 4, 1852–1858 CAS.
- M. Y. Lobanov, M. Y. Suvorina, N. V. Dovidchenko, I. V. Sokolovskiy, A. K. Surin and O. V. Galzitskaya, Bioinformatics, 2013, 29, 1375–1381 CrossRef CAS PubMed.
- K. W. Plaxco, K. T. Simons and D. Baker, J. Mol. Biol., 1998, 277, 985–994 CrossRef CAS PubMed.
- K. F. Fischer and S. Marqusee, J. Mol. Biol., 2000, 302, 701–712 Search PubMed.
- M. M. Gromiha and S. Selvaraj, Curr. Bioinf., 2008, 3, 1–9 CrossRef CAS.
- T. Richa and T. Sivaraman, PLoS One, 2012, 7, e32465 CAS.
- T. Richa, Computational tools for predicting cryptic intermediates and metastable states in the unfolding kinetics of proteins under native conditions, Ph.D. dissertation, SASTRA University, Tamil Nadu, India, 2014 Search PubMed.
- L. Wall, T. Christiansen and J. Orwant, Programming Perl. Sebastopol, O'Reilly Media, Inc., 2000 Search PubMed.
- J. J. Skinner, W. K. Lim, S. Bedard, B. E. Black and S. W. Englander, Protein Sci., 2012, 21, 987–995 CrossRef CAS PubMed.
- J. S. Milne, L. Mayne, H. Roder, A. J. Wand and S. W. Englander, Protein Sci., 1998, 7, 739–745 CrossRef CAS PubMed.
- E. J. Fuentes and A. J. Wand, Biochemistry, 1998, 37, 3687–3698 CrossRef CAS PubMed.
- H. Feng, N.-D. Vu and Y. Bai, J. Mol. Biol., 2005, 346, 345–353 CrossRef CAS PubMed.
- M. Llinas, B. Gillespie, F. W. Dahlquist and S. Marqusee, Nat. Struct. Biol., 1999, 6, 1072–1078 CrossRef CAS PubMed.
- M. Llinas, Investigation of the role of subdomains in protein folding and misfolding. Ph.D. dissertation, University of California, 1999 Search PubMed.
- K. Wuthrich, NMR of proteins and nucleic acids, John Wiley & Sons, New York, 1986 Search PubMed.
- S. L. Mayo and R. L. Baldwin, Science, 1993, 262, 873–876 CAS.
- J. Clarke and A. R. Fersht, Folding Des., 1996, 1, 243–254 CAS.
- N. Bhutani and J. B. Udgaonkar, Protein Sci., 2003, 12, 1719–1731 CrossRef CAS PubMed.
- Y. Bai, J. S. Milne, L. Mayne and S. W. Englander, Proteins, 1994, 20, 4–14 CrossRef CAS PubMed.
- U. Reimer, G. Scherer, M. Drewello, S. Kruber, M. Schutkowski and G. Fischer, J. Mol. Biol., 1998, 279, 449–460 CrossRef CAS PubMed.
- B. M. P. Huyghues-Despointes, J. M. Scholtz and C. N. Pace, Nat. Struct. Biol., 1999, 6, 910–912 CrossRef CAS PubMed.
- L. N. Lin and J. F. Brandts, Biochemistry, 1983, 22, 553–559 CrossRef CAS.
- A. R. Fersht and V. Daggett, Cell, 2000, 108, 573–582 CrossRef.
- F. U. Hartl and M. Hartl-Hayer, Nat. Struct. Mol. Biol., 2009, 16, 574–581 CAS.
- V. N. Uversky, FEBS J., 2010, 277, 2940–2953 CrossRef CAS PubMed.
- Y.-H. Lee and Y. Goto, Biochim. Biophys. Acta, 2012, 1824, 1307–1323 CrossRef CAS PubMed.
- A. D. Miranker and C. M. Dobson, Curr. Opin. Struct. Biol., 1996, 6, 31–42 CrossRef CAS.
- C. M. Dobson, Nature, 2003, 426, 884–890 CrossRef CAS PubMed.
- J. J. Skinner, W. K. Lim, S. Bedard, B. E. Black and S. W. Englander, Protein Sci., 2012, 21, 996–1005 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1: workflow diagram of OneG. Fig. S2: figurative representations of various foldons in cytochrome c unfolding. Fig. S3: figurative representations of various foldons in apocytochrome b562 unfolding. Fig. S4: figurative representations of various foldons in third domain of PDZ unfolding. Fig. S5: figurative representations of various foldons in T4 lysozyme unfolding. See DOI: 10.1039/c4ra04642k |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.