Base-catalyzed retro-Claisen condensation: a convenient esterification of alcohols via C–C bond cleavage of ketones to afford acylating sources†
Feng Xiea,
Fengxia Yana,
Mengmeng Chena and
Min Zhang*ab
aSchool of Chemical & Material Engineering, Jiangnan University, Wuxi 214122, People's Republic of China. E-mail: minzhang@scut.edu.cn; Fax: +86-020-39925999; Tel: +86-13802538592
bSchool of Chemistry & Chemical Engineering, South China University of Technology, 381 Wushan Rd, Guangzhou 510640, People's Republic of China
First published on 23rd June 2014
Abstract
The base-catalyzed esterification of alcohols via retro-Claisen condensation has been demonstrated for the first time. A variety of alcohols including aryl- and heteroaryl methanols as well as aliphatic ones underwent efficient acylation to give the ester products in reasonable to good yields upon isolation. Not only conventional 1,3-diketones but also strong electron-withdrawing group containing acetophenones could serve as the acylating suppliers. The synthetic protocol is operationally simple and adaptable to a broad substrate scope. It complements the existed esterification via the retro-Claisen condensation, and enlarges this benign synthetic methodology.
Introduction
Esters are known as a fundamental class of organic compounds. The ester groups frequently occur in numerous bioactive compounds,1 top-sold marketing medicines2a (e.g., Plavix,2b Concerta,2c Xalatan2d), and synthetic polymers (e.g. proteins and nylons).3 Moreover, esters could serve as interesting building blocks for the preparation of various functionalized products.4 Due to its multiple functions, the development of efficient esterification approaches has long been a subject of synthetic chemists. The conventional syntheses include the Tishchenko5,6 and Fischer esterification,7 trans-esterification,8 and the condensation or coupling reactions of alcohols with carboxylic acids,9 anhydrides,10 acid halides.11 Interestingly, the dehydrogenative esterification established in recent years directly from alcohols demonstrated an alternative approach towards the related end.12 Although these methods are currently the most widely used, many of them suffer a number of drawbacks, such as use of less environmentally benign halogenated reagents, concentrated acid catalysts (i.e., HCl and H2SO4), or expensive ligands and transition-metal catalysts, which could lead to either easy equipment corrosion, or generation of toxic wastes. Therefore, there is a need for the development of acid and transition-metal catalysts free esterification approaches.In recent years, the retro-Claisen condensation has offered a new avenue to access different type of esters. The reaction proceeds via a synergistic process of carbon–carbon (C–C) bond cleavage of ketones and alcohol acylation. Lewis acids such as In(OTf)3, FeCl3 and Fe(OTf)3 have been proven to be efficient catalysts.13 However, to the best of our knowledge, the usage of inexpensive base catalysts for such a transformation has not been explored yet. During the course of our study on α-alkylation of ketones,14 the reaction of pyridin-3-ylmethanol 1a with acetylacetone 2a gave, instead of the anticipated α-alkylated product 3a′, but the ester 3a exclusively (eqn (1)). With this new observation, our further investigations showed that the base (t-BuONa) served as a catalyst in the formation of desired product 3a, while the ruthenium species was not involved in the reaction. Herein, we report a base-catalysed retro-Claisen condensation for the first time, providing a convenient esterification of alcohols via C–C bond cleavage of ketones to afford acylating sources.
 | (1) |
Results and discussion
Firstly, we tried to screen an efficient reaction system. The synthesis of pyridin-3-ylmethyl acetate 3a from 3-pyridin-3-ylmethanol 1a and acetylacetone 2a was chosen as a model reaction to determine different reaction parameters. Initially, the reaction was performed at 120 °C for 12 h in the presence of 20 mol% of t-BuONa. The desired product was detected in 57% yield upon GC analysis (Table 1, entry 1). However, the reaction in absence of base gave only trace of expected product (Table 1, entry 2), indicating the base is an essential catalyst in the formation of product. Subsequently, under the same reaction conditions, replacing t-BuONa with a series of other inorganic and organic bases gave lower product yields (Table 1, entries 3–8). Hence, we chose t-BuONa as the preferred catalyst, other polar and less-polar solvents were showed to be less effective in the formation of 3a comparing to t-amyl alcohol (Table 1, entries 9–11). Gratifyingly, the increase of base loading from 20 mol% to 30 mol% led to an improved product yield (Table 1, entry 12). However, a further increase could not improve the product yield any more (Table 1, entry 13). Similarly, increase or decrease of reaction temperature also failed to result in better reaction efficiency (Table 1, entries 14–15). Finally, prolonging the reaction time from 12 h to 18 h gave a highest yield (Table 1, entry 16). Hence, the optimal reaction conditions can be as indicated in entry 16 of Table 1.
|
|||
|---|---|---|---|
| Entry | Base | Solvent | 3a,b yield% |
| a Reaction conditions: unless otherwise specified, all reactions were carried out without inert gas protection by using 1a (1.2 mmol), 2a (1 mmol), solvent (1 mL), temperature: 120 °C, base (0.2 mmol), reaction time (12 h).b GC yield.c Base (0.3 mmol).d Base (0.5 mmol).e Reaction temperature: 130 °C.f Reaction temperature: 100 °C.g Reaction time: 18 h. | |||
| 1 | t-BuONa | t-Amyl alcohol | 57 |
| 2 | — | t-Amyl alcohol | Trace |
| 3 | K2CO3 | t-Amyl alcohol | 23 |
| 4 | CS2CO3 | t-Amyl alcohol | 34 |
| 5 | KOH | t-Amyl alcohol | 38 |
| 6 | CH3ONa | t-Amyl alcohol | 46 |
| 7 | NEt3 | t-Amyl alcohol | <10 |
| 8 | DABCO | t-Amyl alcohol | 31 |
| 9 | t-BuONa | DMSO | 43 |
| 10 | t-BuONa | DMF | 41 |
| 11 | t-BuONa | Toluene | 52 |
| 12c | t-BuONa | t-Amyl alcohol | 68 |
| 13d | t-BuONa | t-Amyl alcohol | 68 |
| 14e | t-BuONa | t-Amyl alcohol | 68 |
| 15f | t-BuONa | t-Amyl alcohol | 48 |
| 16g | t-BuONa | t-Amyl alcohol | 82 |

With the availability of the optimized reaction conditions in hand, we then examined the scope of the synthetic protocol. Considering the pyridyl group containing esters could often exhibit some interesting biological activities,15 but the esterification of pyridyl methanols by employing retro-Claisen condensation has been scarcely explored. Hence, we firstly focused on the synthesis of pyridylmethyl esters. The reactions of a variety of pyridyl methanols were examined in combination with two representative alkyl and aryl 1,3-diketones (2a, 2b). As shown in Table 2, all the reactions proceeded smoothly and furnished desired esters in moderate to good yields upon isolation (Table 2, entries 1–10). It was found that the substitution patterns of pyridyl methanols slightly affected the product yields. Specifically, the para-pyridyl methanol afforded the corresponding products in higher yields than the meta- and ortho-ones (Table 2, entries 1–3 and 6–8). Meanwhile, the reactions using (2-chloropyridin-3-yl)methanol gave the products in relatively lower yields, which might be attributable to the influence of steric hindrance (Table 2, entries 5, 10). In addition, benzylic alcohols 1f and 1g as well as aliphatic alcohol 1h could also been acylated to afford the desired products in good yields (Table 2, entries 11–14). Noteworthy, the reactions of (2-nitrophenyl)methanol 1f need longer reaction time and elevated reaction temperature to afford desirable yields. It is conceivable that the ortho-NO2 group could significantly decrease the nucleophilicity of alcohol, thus disfavouring the acylation process (Table 2, entries 11–12).
|
|||
|---|---|---|---|
| Entry | Alcohol 1 | Ketone 2 | 3,b yield% |
| a Reaction conditions: unless otherwise specified, all reactions were carried out in presence of 1 (1.2 mmol), 2 (1 mmol), solvent (1 mL), temperature (120 °C), base (0.3 mmol), reaction time (18 h).b Isolated yields.c Temperature: 130 °C, time: 24 h. | |||
| 1 | 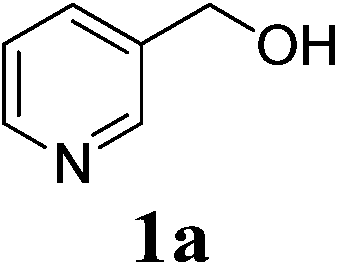 |
2a: R1 = Me | 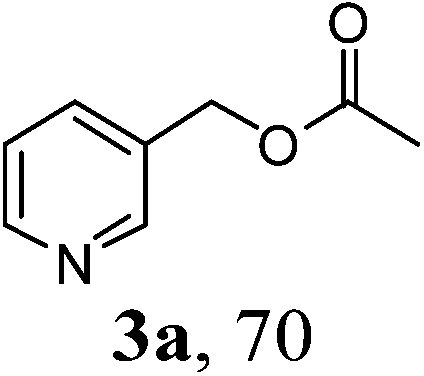 |
| 2 |  |
2a | 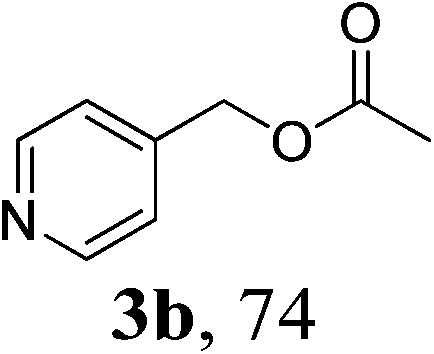 |
| 3 |  |
2a | 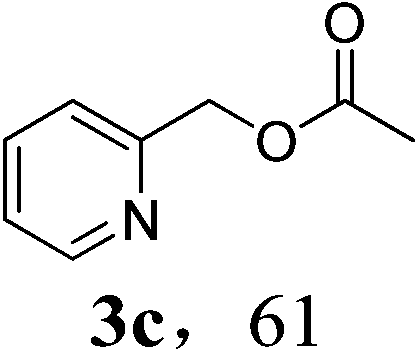 |
| 4 | 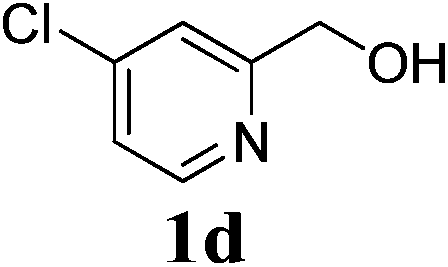 |
2a | 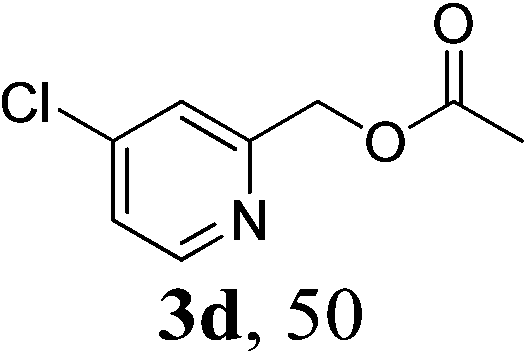 |
| 5 |  |
2a |  |
| 6 | 1a | 2b: R1 = Ph |  |
| 7 | 1b | 2b |  |
| 8 | 1c | 2b |  |
| 9 | 1d | 2b |  |
| 10 | 1e | 2b | 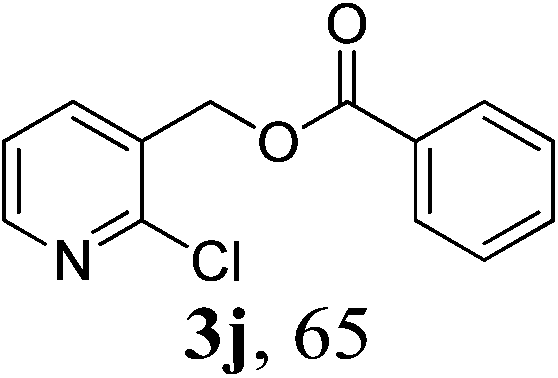 |
| 11c |  |
2a |  |
| 12c | 1f | 2b |  |
| 13 |  |
2b |  |
| 14 |  |
2b |  |

Next, we turned our attention to employ less-reactive acetophenones for the synthetic protocol. Firstly 1-(4-nitrophenyl)ethanone 3a in combination with a variety of alcohols was tested. As shown in Table 3, all the reactions underwent efficiently to afford desired products in reasonable to good isolated yields (Table 3, entries 1–8). Simple benzylic alcohol 1g and aliphatic ones 1j and 1h were proven to be effective coupling partners, giving desired products in good yields (Table 3, entries 1, 3 and 4). Moreover, chloride-containing alcohols (1i, 1k) as well as pyridyl methanols (1a, 1b and 1d) could also be transformed into the corresponding esters in reasonable yields (Table 3, entries 2 and 5–8). However, the reactions of acetophenone 3b, 1-(4-methoxyphenyl)ethanone 3c and 1-(4-fluorophenyl)ethanone 3d failed to yield even trace of expected products (Table 3, entries 9–11), attributing to the acetophenones lacking an strong electron-withdrawing group disfavor of the C–C bond cleavage between the carbonyl group and the α-position.
|
|||
|---|---|---|---|
| Entry | Alcohol 1 | Ketone 3 | 4,b yield% |
| a Reaction conditions: unless otherwise specified, all reactions were carried out without inert gas protection by using 1 (1.2 mmol), 3 (1 mmol), solvent (1 mL), temperature (120 °C), base (0.3 mmol), reaction time (18 h).b Yields of isolated pure product.c Temperature: 130 °C, reaction time: 24 h. | |||
| 1 | 1g | 3a | 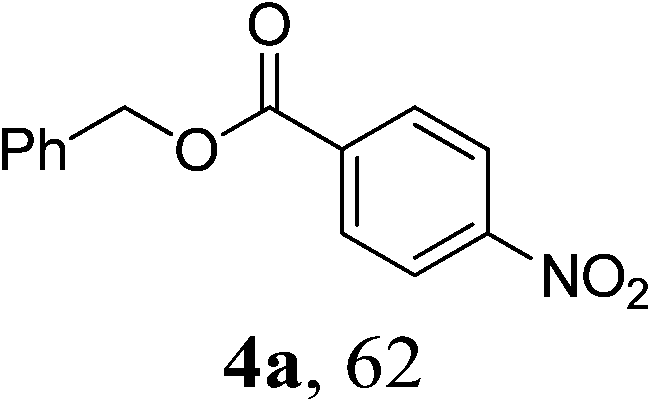 |
| 2 | 1i | 3a |  |
| 3 | 1j | 3a |  |
| 4 | 1h | 3a | 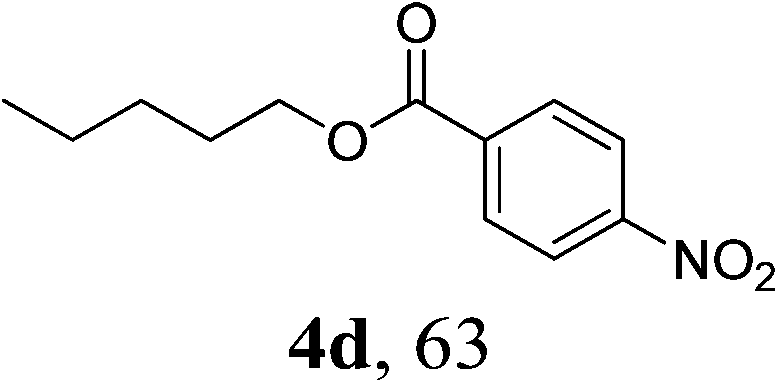 |
| 5c | 1k | 3a |  |
| 6 | 1a | 3a |  |
| 7 | 1b | 3a |  |
| 8 | 1d | 3a |  |
| 9 | 1g | 3b | — |
| 10 | 1g | 3c | — |
| 11 | 1g | 3d | — |

Finally, unsymmetrical 1-phenylbutane-1,3-dione 2c was used in combination with pyridin-2-ylmethanol 1c to investigate the reaction chemoselectivity, which could lead to two possible isomers 3c and 3h. Interestingly, except for the generation acetophenone 3b, ester 3c and 3h were observed in a ratio of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Scheme 1). The selectivity is associated with the sterically less hindered acyl moiety favor the acylation of alcohol.
:1 (Scheme 1). The selectivity is associated with the sterically less hindered acyl moiety favor the acylation of alcohol.
 | ||
| Scheme 1 The esterification of dissymmetric 1,3-diketone and alcohols. | ||
Based on the retro-Claisen condensation,16 a plausible pathway for the generation of esters from alcohols and ketones is depicted in Scheme 2. The reaction initiated with t-BuONa-induced deprotonation of alcohol 1 to form alkoxy anion A, A undergoes a nucleophilic attack to the carbonyl group of ketone and affords intermediate B. Then, by abstracting a proton from the in situ formed t-BuOH, the thermodynamic favorable C–C bond cleavage of B in a retro-Aldol manner would afford the desired product, regenerate the t-BuONa catalyst and release the side product R3Me, respectively.
 | ||
| Scheme 2 Proposed mechanism for the esterification of keones. | ||
Conclusions
In summary, we have demonstrated a base-catalyzed esterification of alcohols via retro-Claisen condensation for the first time. A variety of alcohols including aryl- and heteroaryl methanols as well as aliphatic ones underwent efficient acylation to give the ester products in reasonable to good isolated yields. Not only conventional 1,3-diketones but also strong electron-withdrawing group containing acetophenones could serve as the acylating suppliers. The synthetic protocol is operationally simple and adaptable to a broad substrate scope. It complements the existed esterification via the retro-Claisen condensation, and enlarges this benign synthetic methodology.Experimental
General information
All the obtained products were characterized by melting points (m.p), 1H-NMR, 13C-NMR infrared spectra (IR), the NMR spectra of the known compounds were found to be identical with the ones reported in the literatures. Additionally, the solid and new products were further determined by melting points and mass spectra (MS), respectively. 1H-NMR and 13C-NMR spectra were obtained on a Bruker Avance 400 MHz NMR spectrometer; mass spectra were recorded on Trace DSQ GC/MS. Melting points were measured on an Electrothemal SGW-X4 microscopy digital melting point apparatus and are uncorrected; IR spectra were recorded on a Brucker Vector 22 spectrometer; chemical shifts were reported in parts per million (ppm, δ) downfield from tetramethylsilane. Proton coupling patterns are described as singlet (s), doublet (d), triplet (t), multiplet (m); TLC was performed using commercially prepared 100–400 mesh silica gel plates (GF254), and visualization was effected at 254 nm; all the reagents were purchased from commercial sources (J&K Chemic, TCI, Fluka, Acros, SCRC), and used without further purification.Typical procedure for the synthesis of 3-pyridylmethyl acetate 3a
t-BuONa (0.028 g, 0.3 mmol), 3-(hydroxymethyl)pyridine 1a (0.131 g, 1.2 mmol), acetylacetone 2a (0.1 g, 1 mmol) and t-amyl alcohol (1 mL) were added successively to a Schlenk tube, it was then closed and the resulting mixture was heated at 120 °C for 18 h in an oil bath. After cooling down to room temperature, the reaction mixture was then filtered and concentrated under vacuum, the obtained crude product was directly purified by preparative TLC on silica, eluting with petroleum ether (60–90 °C)–ethyl acetate (3:1) to give 3-pyridylmethyl acetate 3a as a yellow oil (0.110 g, 75%).
Analytic data of the obtained compounds
Acknowledgements
The authors are grateful to the funds of the “National Natural Science Foundation of China (21101080)”, “Natural Science Foundation of Jiangsu Province (BK2011144)”, “Fundamental Research Funds for the Central Universities of China (2014ZZ0047)”, and “Distinguished talent program of SCUT”.Notes and references
- For selected examples, see:
(a) E. Armani, G. Amari, A. Rizzi, R. D. Fanti, E. Ghidini, C. Capaldi, L. Carzaniga, P. Caruso, M. Guala, I. Peretto, E. La Porta, P. T. Bolzoni, F. Facchinetti, C. Carnini, N. Moretto, R. Patacchini, F. Bassani, V. Cenacchi, R. Volta, F. Amadei, S. Capacchi, M. Delcanale, P. Puccini, S. Catinella, M. Civelli and G. Villetti, J. Med. Chem., 2014, 57, 793–816 CrossRef CAS PubMed
; (b) A. Trotier-Faurion, S. Dezard, F. Taran, V. Valayannopoulos, P. de Lonlay and A. Mabondzo, J. Med. Chem., 2013, 56, 5173–5181 CrossRef CAS PubMed
-
(a) Esterification, ed. J. Otera, Wiley-VCH, Weinheim, 2003 Search PubMed
- M. Aden, V. Mamuschkin, A. Olowinsky and S. Glaser, J. Appl. Polym. Sci., 2014, 131, 40073 CrossRef
-
(a) X. Sun, Z. Song, H. Li and C. Sun, Chem.–Eur. J., 2013, 19, 17589–17594 CrossRef CAS PubMed
-
(a) J. Li, J. Shi, H. Han, Z. Guo, H. Tong, X. Wei, D. Liu and M. F. Lappert, Organometallics, 2013, 32, 3721–3727 CrossRef CAS
-
(a) K. Rajesh and H. Berke, Adv. Synth. Catal., 2013, 355, 901–906 CrossRef CAS
- J. P. Guzowski, E. J. Delaney, M. J. Humora, E. Irdam, W. F. Kiesman, A. Kwok and A. D. Moran, Org. Process Res. Dev., 2012, 16, 232–239 CrossRef CAS
-
(a) N. Degirmenbasi, N. Boz and D. M. Kalyon, Appl. Catal., B, 2014, 150, 147–156 CrossRef PubMed
-
(a) N. Nowrouzi, A. M. Mehranpour and J. A. Rad, Tetrahedron, 2010, 66, 9596–9601 CrossRef CAS PubMed
-
(a) M. Moghadam, S. Tangestaninejad, V. Mirkhani, I. Mohammadpoor-Baltork, M. Babaghanbari, M. Zarea, L. Shariati and S. A. Taghavi, J. Iran. Chem. Soc., 2009, 6, 523–532 CrossRef CAS
-
(a) M. A. Pasha, M. B. M. Reddy and K. Manjula, Eur. J. Chem., 2010, 1, 385–387 CrossRef CAS PubMed
-
(a) J.-B. Feng, J.-L. Gong, Q. Li and X.-F. Wu, Tetrahedron Lett., 2014, 55, 1657–1659 CrossRef CAS PubMed
-
(a) C. B. Rao, D. C. Rao, D. C. Babu and Y. Venkateswarlu, Eur. J. Org. Chem., 2010, 2855–2859 CrossRef CAS
- F.-X. Yan, M. Zhang, X.-T. Wang, F. Xie, M.-M. Chen and H.-F. Jiang, Tetrahedron, 2014, 70, 1193–1198 CrossRef CAS PubMed
-
(a) I. Im, E. S. Lee, S. J. Choi, J.-Y. Lee and Y.-C. Kim, Bioorg. Med. Chem. Lett., 2009, 19, 3632–3636 CrossRef CAS PubMed
-
(a) M. Jukic, D. Sterk and Z. Casar, Curr. Org. Synth., 2012, 9, 488–512 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra04618h |
| This journal is © The Royal Society of Chemistry 2014 |