
Computational study on redox-switchable second-order nonlinear optical properties of ferrocene-tetrathiafulvalene hybrid†
Chun-Guang Liuab,
Ming-Li Gaob and
Zhi-Jian Wu‡
*a
aState Key Laboratory of Rare Earth Resource Utilization, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, 130022, P. R. China. E-mail: zjwu@ciac.ac.cn; Fax: +86-431-85698041; Tel: +86-431-85262035
bCollege of Chemical Engineering, Northeast Dianli University, Jilin City, 132012, P. R. China
First published on 13th August 2014
Abstract
Redox-switchable second-order nonlinear optical (NLO) responses of a series of ferrocene-tetrathiafulvalene (Fc–TTF) hybrids have been studied based on density functional theory calculations. The hyper-Rayleigh scattering (HRS) responses as well as the dynamic (λ = 1064 nm) HRS hyperpolarizabilities have been calculated in the gas phase within the T convention. The electron-correlation effects have been investigated. The long-range corrected LC-BLYP and wB97X-D functionals provide satisfactory results. The electron donor strength of the Fc–TTF in a donor-π-conjugated-acceptor structure has been assessed. The results indicate that the Fc unit does not play the role of the electron donor in the Fc–TTF unit. Because the Fc–TTF hybrid unit is a multistep redox center, the one- and two-electron-oxidized processes have been considered to control the second-order NLO responses. For a known Fc–TTF hybrid, one-electron-oxidization leads to a significant increase of the HRS hyperpolarizability, while the calculated HRS hyperpolarizabilities are not affected by the two-electron-oxidization according to our DFT calculations. Interestingly, in another system the two-electron-oxidization significantly enhances the HRS hyperpolarizability, and the one-electron-oxidization does not largely affect the HRS hyperpolarizability.
1. Introduction
Because of the appealing potential applications in optical communication or data storage, the development of materials exhibiting switchable nonlinear optical (NLO) properties have motivated a number of studies during the past two decades.1–3 The reported methods for the switching of NLO properties include redox,4–9 protonation/deprotonation,10–12 photoisomerization,13,14 boron site reduction,15 ion induction in solution,16,17 etc. In order to obtain an effective redox-switching of second-order NLO responses (the molecular first hyperpolarizability β), several questions should be considered. Such as, the stability of two forms relative to the switching of ‘on’ and ‘off’ is important because they can preferably be switched from one to other in an easily controlled way and the fast response time.7 Molecular system with redox-switchable NLO responses is particularly attractive because the oxidation or reduction (removal or addition of electrons) enables fine control of their NLO properties. Although a large variety of redox-active molecular systems have been reported, the excellent redox center possessing the ability of reversible modification of second-order NLO responses is rare.As illustrated in Chart 1, the redox-active tetrathiafulvalene (TTF) unit is able to exist in three different stable redox states (TTF, the radical cation TTF·+, and dication TTF2+), and the oxidation to the radical cation and dication occurs sequentially and reversibly at low potentials.18–24 Due to the 14 π-electron structure, the TTF unit is lack of a π-electron cyclic conjugation, and thus is nonaromatic according to Hückel rule. But the radical cation and dication with one and two 6 π-electrons are aromatic in the Hückel sense, and thus both oxidized species are the thermodynamically stable. On the basis of the present points, the TTF unit is an ideal model for the redox switching of NLO responses. A series of redox-switchable second-order NLO molecules containing TTF unit have been studied experimentally and theoretically.13,14,25–27 A large contrast in their second-order NLO responses has been obtained in TTF → TTF·+ oxidization. By contrast, the TTF·+ → TTF2+oxidization does not largely affect their second-order NLO responses.13,14 It is well-known that the neutral TTF unit possesses a bent geometry, and the bent TTF unit changes to a fully planar geometry in the one-electron-oxidized process. Thus the large contrast in second-order NLO responses in TTF → TTF·+ oxidization is relative to a significant geometrical change of the TTF unit.
 | ||
| Chart 1 Sequential and reversible oxidization of TTF unit. | ||
The ferrocene (Fc) is another typical redox center for reversibly modifying second-order NLO responses because it is easy to prepare and energetically convenient FeII/FeIII couple.28–36 It has been demonstrated that the oxidization of FeII to FeIII caused the electron absorption spectra (charge transfer transition) of the FeII and FeIII derivatives are decisively different from each other in a chromophore containing octamethylferrocenyl and nitro-thiophenyl groups.37 According to sum-over-state description, any modification of the absorption spectrum of a molecule would alter the first hyperpolarizability. The hyper-Rayleigh scattering (HRS) experiments indicated that the FeII → FeIII oxidization led to the first hyperpolarizability to decrease very substantially.
Due to the coexistence of 3d and π-spin electrons in the same molecule, the preparation of Fc–TTF hybrid has attracted considerable attention in the past three decades.38–46 Several Fc–TTF and Fc–metalladithiolene hybrids have been reported, and their multistep redox properties have been investigated by cyclic voltammetry. However, their redox-switchable second-order NLO properties have not been explored. The molecule existing in two stable states to switching of second-order NLO response has been regarded as a good candidate for binary digital architecture because the redox interconversion between the two stable states matched the binary zero and one. In order to obtain a smaller molecule-based information-processing device component, the higher-order digit representation (such as ternary, senary, etc.) may be much superiority relative to binary.47 Toward such goals, molecule must be able to exist in more than two stable and independently addressable states.48 The Fc–TTF hybrid with multistep redox properties provides a very good basis for development of device component with high-order digit representation.
Here, we report a detailed theoretical study on a series of Fc–TTF hybrids with the aim of optimization of second-order NLO property by an effective combination of the Fc and TTF unit in the same molecule and describing the relationship between multistep redox properties and second-order NLO properties.
2. Computational details
All calculations were performed using Gaussian09 program.49 The molecular structures were optimized and characterized as energy minima at B3LYP50–52/6-31g (d) level (LANL2DZ basis sets53–55 for metal atom). The frequency-dependent hyperpolarizability was calculated by using time-dependent Hatree Fock (TDHF) and TD density functional theory (DFT) method at 1064 nm in this work.56–60 The reliability of DFT with conventional exchange correlation (XC) functionals to evaluate the molecular hyperpolarizability has been questioned in a large number of studies. A correct description of the asymptotic behavior of the XC potential may be a prerequisite for the correct description of the first hyperpolarizability. Castet and Champagne have assessed a series of DFT XC functionals for evaluating the multipolar contributions to the first hyperpolarizability (the HRS responses) of some reference molecules.61 They concluded that the best functionals were LC-BLYP,62 M05-2X,63,64 and M06-2X65 for description of the HRS hyperpolarizability of their reference molecules. In the present paper, the six different functionals including BHandHLPY,66 wB97X-D,67,68 CAM-B3LYP,69 LC-BLYP, M05-2X, and M06-2X have been employed to calculated the frequency-dependent hyperpolarizability. In order to evaluate the electron correlation effect of these functionals, as a reference the Hatree Fock (HF) method without electron-correlation consideration also has been carried out to calculate the frequency-dependent hyperpolarizability in this work. The basis sets containing diffuse and polarization functions are necessary for accurate predictions of hyperpolarizability according to many literatures. In the present paper, the 6-31+g(d) basis sets with a good compromise between accuracy and computational costs for large molecular system have been employed for the first hyperpolarizability calculations (LANL2DZ basis sets for metal atom).The available technology for measuring second-order NLO response is mainly HRS and electric field induced second harmonic generation (EFISHG) experimentally. HRS is the only experimental method to allow measuring the second-order NLO response of the charged species. Thus, we focused the HRS response in this work. Theoretically, Champagne et al. developed an effective method to estimate the HRS responses.70–77 According to Bersohn's expression, the second-order NLO response βHRS(−2ω; ω, ω) can be extracted from HRS data by using the following equation:78,79
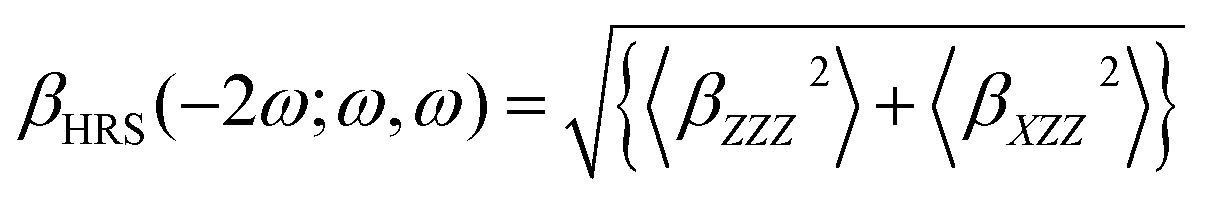 | (1) |
The depolarization ratio (DR), which is associated with the shape of the NLO-phore, given by:
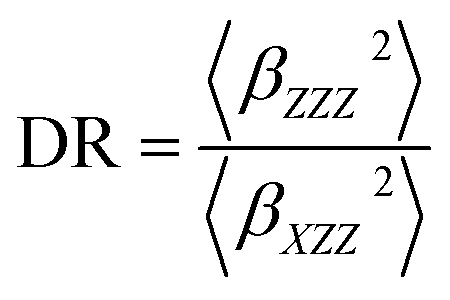 | (2) |
In order to get more insights of the origin of second-order NLO responses, the TDDFT method has been employed to calculate the nature of excited state. The excited energy, oscillator strengths, and associated major contributions of the orbital transitions for the studied molecule were determined at the B3LYP level using 6-31g(d) basis sets (LANL2DZ basis sets for metal atom). TDDFT is a usefully accurate approach for many applications,83 especially, low-lying single excitations.
3. Results and discussion
3.1. The donor strength of Fc–TTF hybrid
The typical second-order NLO chromophores have mostly focused on the donor-π conjugated-acceptor (D-π-A) structure. The Fc and TTF units are both the electron donor groups. The donor strength of the Fc unit is only comparable to the organic methoxyphenyl group according to experimental and theoretical studies.36 And the donor strength of the TTF unit is slightly smaller than that of the typical N,N-bis-(4-methoxyphenyl)phenyl-amino donor based on our previous works.13 However, the donor strength of the combination of Fc and TTF units has not been reported and is thus worthy of exploration. Firstly, an array model of the two donor groups in a same molecule should be considered. Several patterns of covalently bonded Fc and TTF units have been reported. Recently, a novel Fc–TTF hybrid, FcS4TTF(R)2 (R = CF3 and SMe (H)) (see Fig. 1), has been synthesized successfully, where the Fc and TTF units are linked to each other through an multisulfur π-conjugated bridge.84 This compound is unique for designing the second-order NLO molecular system because the π-conjugated bridge would improve the coupling between the Fc and TTF units, and thus enhance their second-order NLO responses.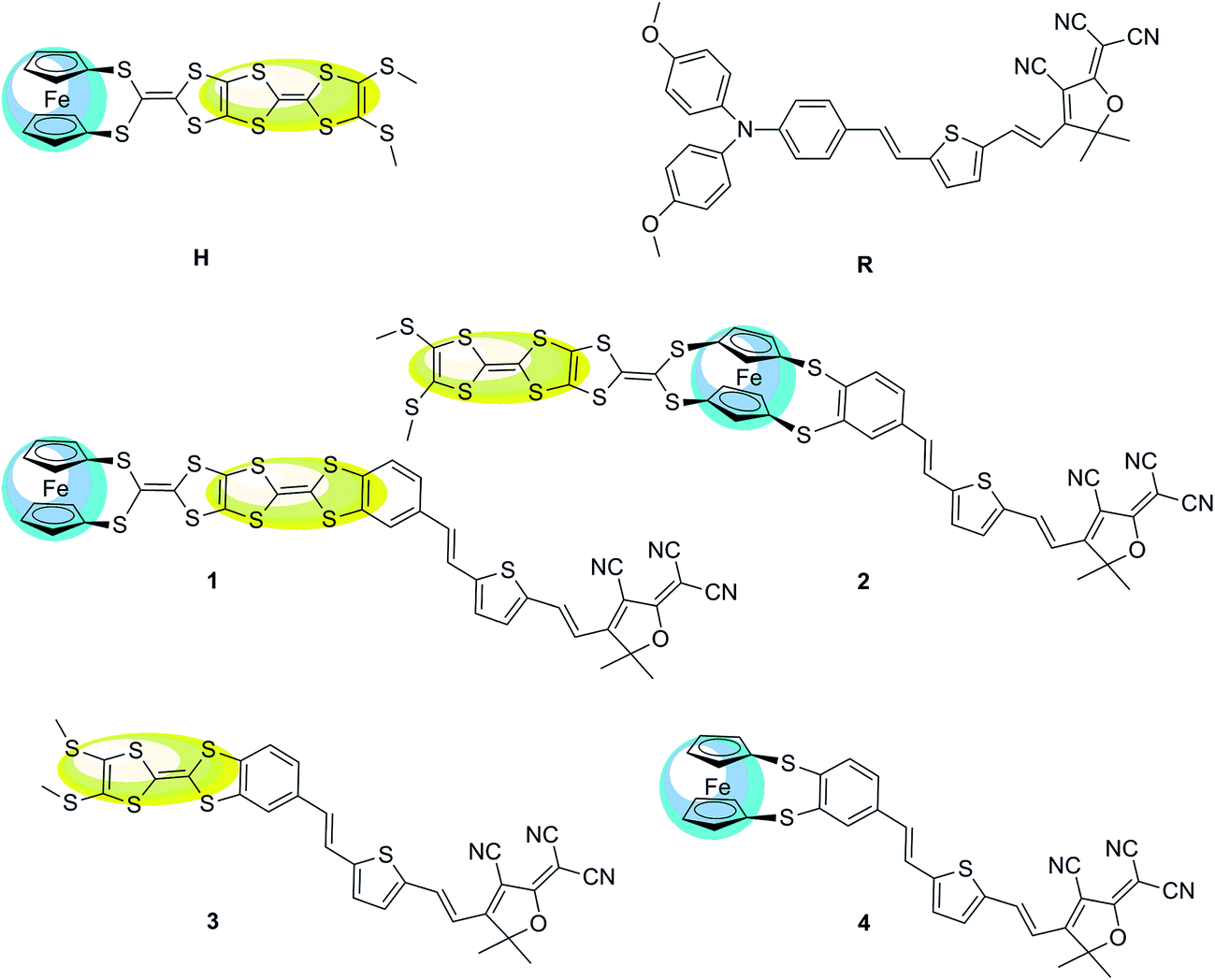 | ||
| Fig. 1 Structural formula of the Fc–TTF hybrid H, reference system R, and their derivatives 1, 2, 3, and 4 (the yellow and blue color highlight TTF and Fc unit, respectively). | ||
Due to the Fc and TTF units are both the electron donor, we will introduce a π-conjugated bridge and an electron acceptor to design a new molecular system with D-π-A structure. A compound containing the key N,N-bis-(4-methoxyphenyl)phenyl-amino donor, 2,5-divinylthienyl π-conjugated bridge, and the 2-dicyanomethylen-3-cyano-4-methyl-5-phenyl-5-trifluoromethyl-2,5-dihydrofuran acceptor (R) (see Fig. 1) has been chosen for designing the D-π-A structure because the second-order NLO property of this compound has been investigated detailedly and broadly.85 It has been demonstrated that this compound has a 2-fold improvement in the molecular first hyperpolarizability and 3-fold macroscopic response than the simple triphenylamino analogue.86
As shown in Fig. 1, the donor moiety of R (N,N-bis-(4-methoxyphenyl)phenyl-amino) has been replaced with the Fc–TTF group (FcS4TTF(SMe)2), and thus we have systems 1 and 2, where the π-A moiety of R was attached to the Fc–TTF group at TTF (1) and Fc (2) end, respectively. The frequency-dependent hyperpolarizabilities βHRS(−2ω; ω, ω) of H, 1, and 2 have been calculated at HF, LC-BLYP, wB97X-D, CAM-B3LYP, BHandHLYP, M05-2X, and M06-2X levels using 6-31+g(d) basis sets (LANL2DZ basis sets for metal atom) in this work. The calculated βHRS(−2ω; ω, ω) value are given in Table 1. In order to estimate the electron correlation effects of different functionals, the ratio between the DFT-derived βHRS(−2ω; ω, ω) values and the corresponding HF values also has been reported in Table 1. It can be found that the βHRS(DFT)/βHRS(HF) ratio depends on the functional and differs from one molecule to another. Compared to the HF reference, all DFT calculations lead to an increase of the βHRS(−2ω; ω, ω) values for all three compounds. For H and 2, the βHRS(DFT)/βHRS(HF) ratio is controlled in a reasonable range, where the ratio ranges from 1.40 to 3.10 in H and 1.46 to 4.84 in 2. However, a significant difference is obtained in the case of 1, where the fluctuation of βHRS(DFT)/βHRS(HF) ratio is very large (ranging from 1.54 to 852.93). This result confirms that the electron correlation effects of four functionals (CAM-B3LYP, BHandHLYP, M05-2X, and M06-2X) fail to describe the first hyperpolarizability because of a large fluctuation of βHRS(DFT)/βHRS(HF) ratio along with one molecule to another. By contrast, two functionals LC-BLYP and wB97X-D provide a rational result for the first hyperpolarizability calculation of 1, where the βHRS(DFT)/βHRS(HF) ratio is 1.54 and 4.81, respectively. Especially the LC-BLYP functional, the calculated βHRS(DFT)/βHRS(HF) ratios of H, 1, and 2 are in a narrow range (1.85 for H, 1.54 for 1, and 1.46 for 2). This result indicates that the major part of electron correlation effects is included at the LC-BLYP level for the three systems.
| Compounds | Method | βHRS | βHRS(DFT)/βHRS(HF) | DR |
|---|---|---|---|---|
| a 1 a.u. = 3.62 × 10−42 m4 V−1 = 8.64 × 10−33 esu. | ||||
| H | HF | 80 | — | 4.97 |
| LC-BLYP | 148 | 1.85 | 2.64 | |
| wB97X-D | 190 | 2.38 | 2.18 | |
| CAM-B3LYP | 177 | 2.21 | 2.16 | |
| BHandHLYP | 112 | 1.40 | 1.59 | |
| M05-2X | 213 | 2.66 | 2.26 | |
| M06-2X | 248 | 3.10 | 3.08 | |
| 1 | HF | 77![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 088 088 |
— | 4.95 |
| LC-BLYP | 119182 |
1.54 | 4.99 | |
| wB97X-D | 371371 |
4.81 | 5.00 | |
| CAM-B3LYP | 1609449 |
20.88 | 5.00 | |
| BHandHLYP | 65901109 |
852.93 | 5.01 | |
| M05-2X | 21752336 |
282.17 | 5.00 | |
| M06-2X | 3097211 |
40.09 | 5.00 | |
| 2 | HF | 47448 |
— | 4.91 |
| LC-BLYP | 69202 |
1.46 | 4.98 | |
| wB97X-D | 137184 |
2.89 | 5.00 | |
| CAM-B3LYP | 208214 |
4.39 | 5.00 | |
| BHandHLYP | 215506 |
4.54 | 5.00 | |
| M05-2X | 201306 |
4.24 | 5.00 | |
| M06-2X | 229712 |
4.84 | 5.00 | |
| 3 | HF | 77679 |
— | 4.95 |
| LC-BLYP | 121826 |
1.57 | 4.99 | |
| wB97X-D | 423339 |
5.45 | 5.00 | |
| CAM-B3LYP | 3608947 |
46.46 | 5.00 | |
| BHandHLYP | 3021202 |
38.89 | 5.00 | |
| M05-2X | 2328114 |
29.97 | 5.00 | |
| M06-2X | 1457492 |
18.76 | 5.00 | |
| 4 | HF | 52042 |
— | 4.91 |
| LC-BLYP | 77112 |
1.48 | 4.97 | |
| wB97X-D | 159483 |
3.06 | 4.99 | |
| CAM-B3LYP | 253183 |
4.86 | 5.00 | |
| BHandHLYP | 203016 |
3.90 | 5.00 | |
| M05-2X | 246775 |
4.74 | 5.00 | |
| M06-2X | 288005 |
5.53 | 5.00 | |
All the first hyperpolarizability calculations supported that (i) introduction of the π-A moiety into the studied molecular system leads to a substantial enhancement of the βHRS(−2ω; ω, ω) value (1 vs. H or 2 vs. H); (ii) the second-order NLO responses are sensitive to the link location between D and π-A moieties (1 vs. 2), where the system 1 (the π-A moiety of R was attached to the Fc–TTF group at TTF end) possesses the largest first hyperpolarizability among the three compounds. According to LC-BLYP calculation, the βHRS(−2ω; ω, ω) value of 1 is about 2 and 800 times as large as that of 2 and H, respectively. (iii) The DR increases to the 5 due to introduction of the π-A moiety, which demonstrates the NLO-phore of 1 and 2 would display ideal one-dimensional structures.
On the basis of the complex sum-over-states expression, Oudar and Chemla87,88 established a simple link between the molecular hyperpolarizability and a low-lying energy charge transfer transition through the two-level model,
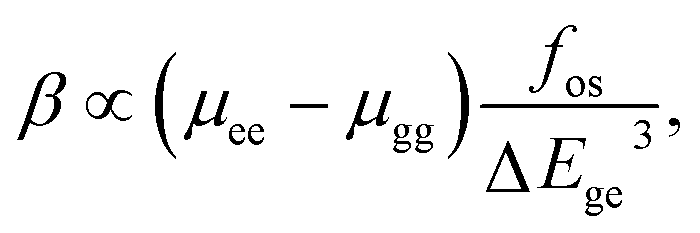 | (3) |
The TDDFT method has been carried out to calculate the excited state of the system 1. The TDDFT calculated excited energy, oscillator strength, and associated orbital transition of the crucial excited state have been listed in Table 2. The crucial excited state is defined as the lowest optically allowed excited state with substantial oscillator strength. Our TDDFT calculations show that the crucial excited state of the system 1 is the third excited state. It contains the HOMO−1 → LUMO (45%), HOMO → LUMO (24%), and HOMO−2 → LUMO (24%) orbital transitions (see Table 2). The associated frontier molecular orbitals (FMOs) of the crucial excited state also have been listed in Table 2. It can be found that the HOMO is mainly localized on the TTF unit, and the LUMO is mainly localized on the electron acceptor end, which indicates that both FMOs are separated completely. This result indicates that the HOMO → LUMO orbital transition would give a significant charge transfer (CT) from TTF unit to the electron acceptor end. However, due to the hardly any overlap between the two orbitals, such electron transition does not largely contribute to the crucial excited state (24%). By contrast, the major contribution arises from the HOMO−1 → LUMO orbital transition (45%) according to our TDDFT calculations. As shown in Table 2, the HOMO−1 is delocalized over the whole molecule, combination of the electron density distribution in the LUMO, which indicates that the crucial excitation of the system 1 can be viewed as a CT transition from the TTF unit to electron acceptor end. We also note that electron density on the Fc unit is very small in the series of occupied molecular orbitals (HOMO, HOMO−1, and HOMO−2). This indicates that the Fc unit does not display the role of electron donor in the system 1.
| Compound | Excited state | fos | E (eV) | Major contributions |
|---|---|---|---|---|
| 1 | 3 | 1.97 | 2.38 | HOMO−1 → LUMO (45%) |
| HOMO → LUMO (24%) | ||||
| HOMO−2 → LUMO (24%) | ||||
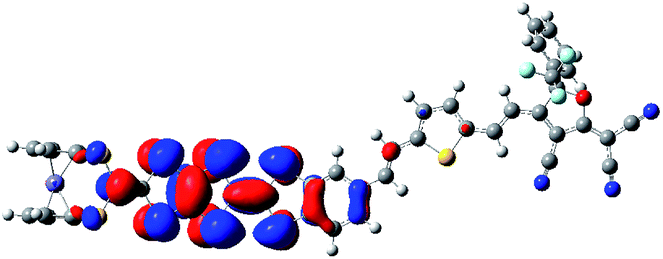 |
 |
|||
| HOMO | LUMO | |||
 |
 |
|||
| HOMO−1 | HOMO−2 | |||
In order to evaluate the donor strength of the Fc–TTF unit in the system 1 further, two new molecular systems 3 and 4 also have been considered in this work. Where each system only includes Fc or TTF donor (see Fig. 1). The frequency-dependent hyperpolarizabilities βHRS(−2ω; ω, ω) of 3 and 4 also have been calculated at the same levels, and are given in Table 1. As expected, all DFT calculations overestimate frequency-dependent hyperpolarizability βHRS(−2ω; ω,ω) value of both systems with respect to the HF reference. For the system 3, the LC-BLYP and wB97X-D functionals satisfactorily describes the βHRS(−2ω; ω, ω) value, while CAM-B3LYP, BHandHLYP, M05-2X, and M06-2X functionals are failed again because of the overestimate of βHRS(−2ω; ω, ω) value with respect to the HF reference. For the system 4, all the DFT calculations provide the βHRS(DFT)/βHRS(HF) ratio ranging from the 1.48 to the 5.53. Within the series of functionals considered in this study, the best functional for describing the second-order NLO response of the five systems appears to be LC-BLYP functional because of a stable βHRS(DFT)/βHRS(HF) ratio from one molecule to another (1.85 for H, 1.54 for 1, 1.46 for 2, 1.57 for 3, and 1.48 for 4). The wB97X-D functional also provide a well result with a βHRS(DFT)/βHRS(HF) ratio ranging from 2.35 to 5.45 along with the series of systems.
The LC-BLYP-derived βHRS(−2ω; ω, ω) values for 1 and 3 are 119182 and 121826 a.u., respectively. It is clearly show that the second-order NLO responses of 1 and 3 are very similar to each other. This result indicates that the second-order NLO properties of both systems are not sensitive to the presence of Fc unit (see Fig. 1). The Fc unit does not display the role of electron donor in the system 1, which agrees well with the TDDFT calculations.
3.2. Redox-switchable second-order NLO response
The electrochemical properties of H have been reported. Cyclic voltammetry showed a series of reversible oxidation waves.84 The first redox potential Eo/ox1 is at 0.11 V vs. Fc+/Fc (−0.33 V vs. normal hydrogen electrode (NHE)), the second redox potential Eo/ox2 appears at 0.30 V vs. Fc+/Fc (−0.14 V vs. NHE). And the first and second oxidation center of H has been assigned to the TTF and Fc moieties, respectively. We now estimate the cyclic voltammetric data of H based on the present DFT calculation. The theoretical estimation of the redox potential of a given molecular system requires determination of the free energy change of the half-reactions, which is represented by the following thermodynamic cycle (4)
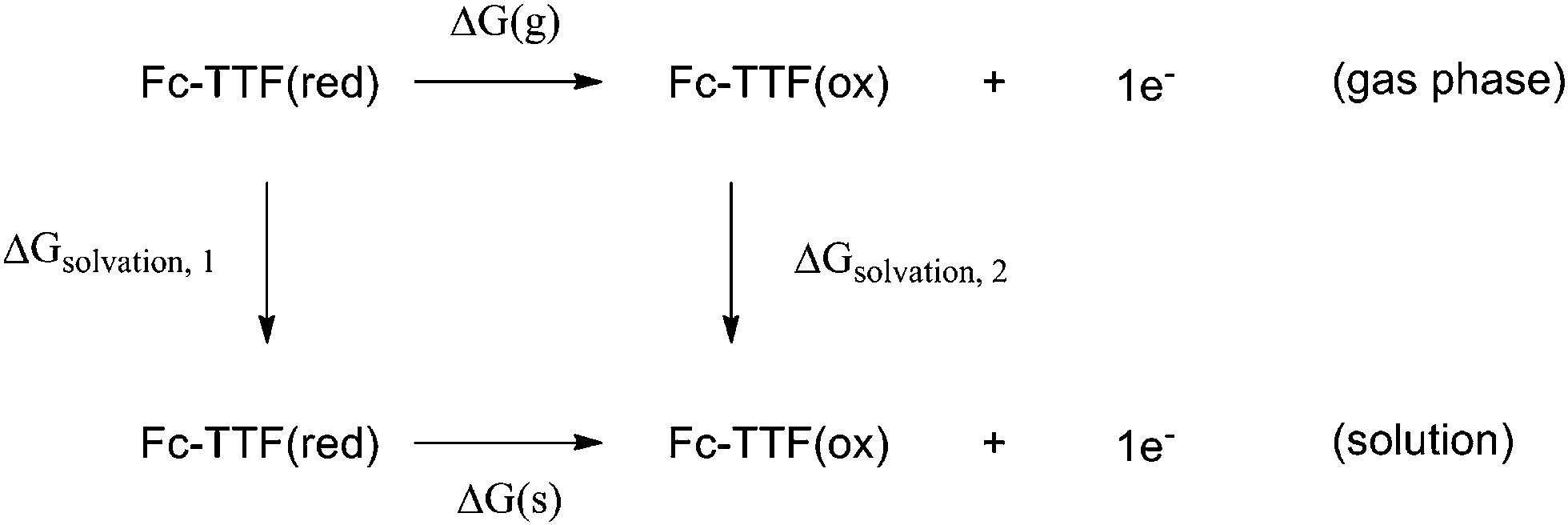 | (4) |
The ΔG(g) term represents the free energy associated with the oxidization of H in the gas phase; the next terms ΔGsolvation, 1 and ΔGsolvation, 2 correspond to the solvation free energies of the reduced and oxidized forms. Finally ΔG(s) term is the free energy of the oxidization process in solution. The molecular geometries of H and its one- and two-electron-oxidized (2H+oxidized and H2+oxidized) species have been optimized at B3LYP/6-31g(d) levels in this work (LANL2DZ basis sets for metal atom). On the basis of the optimized geometries, the free energies of the species corresponding to the thermodynamic cycle have been calculated at the same level. The solvation free energies have been taken into account via the self-consistent reaction field (SCRF) method, using the integral equation formalism polarizable continuum model (IEFPCM) solvent model.89 The SMD solvation model90 proposed by Truhalr and coworkers has been performed for computing solvation free energies. A dielectric constant of acetonitrile has been employed in this work.
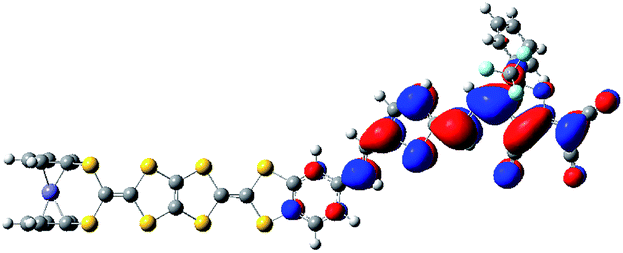
The Fe atom in H is formally in an oxidation state of +2 without unpaired d electrons, giving rise to a ground state with closed-shell configuration (singlet state). Due to removal of an electron from the singlet state, the spin state of the one-electron-oxidized species of H is doublet (an unpaired electron 2H+oxidized). The spin density distribution of 2H+oxidized has been listed in Fig. 2. It can be found that the unpaired electron of 2H+oxidized is mainly localized on the TTF moiety. This indicates that the first oxidization should take place preferentially at the TTF moiety. For the two-electron-oxidized species, the measurement of magnetic property showed that it possessed a diradical character with 3d and π spin.84 And thus the optimized calculations have been carried out based on the broken-symmetry DFT method in this work. Meanwhile, the singlet state of the two-electron-oxidized species (1H2+oxidized) also has been considered. Our DFT calculations show that the most stable species is the antiferromagetic state (antiH2+oxidized). The calculated total energy including the electronic and zero-point corrected energies of antiH2+oxidized is ∼82.32 kJ mol−1 stable than 1H2+oxidized. As shown in Fig. 2, the spin densities of antiH2+oxidized are mainly localized over the Fe center and TTF moiety with antiferromagetic spin alignment (blue, α spin; green, β spin). Combined with the spin density of 2H+oxidized, all results indicate that the second oxidized center should be the Fe center. Taking into account all the above points, our DFT calculations well reproduced the oxidation center of H when compared with the experimental studies.
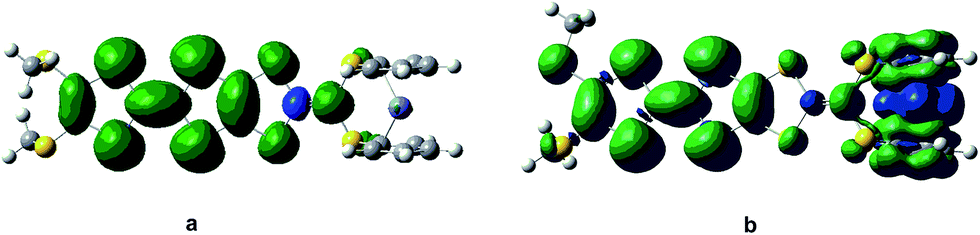 | ||
| Fig. 2 Spin density distribution of H in one- (a) and two-electron-oxidized species (b). | ||
On the basis of the ground-state structure for the series of oxidized species of H, we will reproduce the redox potential of the present studies Fc–TTF hybrid. The half-reaction of normal hydrogen electrode (NHE) is defined as 1/2H2(g) → H+(aq) + e−(g), and the absolute reduction potential of the NHE has been computed to be 4.36 eV (note that the sign of the absolute potential depends on the direction in which the reaction is written: the absolute oxidation potential of the hydrogen electrode is negative while the absolute reduction potential is positive).91 Using this value and these oxidization potentials, the free energies of the two oxidization processes are consequently estimated to be −4.71 and −4.48 V according to experimental studies.84 Our DFT calculated energies is −4.70 and −4.49 V, which is well in agreement with the experimental values.
The frequency-dependent hyperpolarizability of 2H+oxidized and antiH2+oxidized also has been calculated at HF, LC-BLYP, wB97X-D, CAM-B3LYP, BHandHLYP, M05-2X, and M06-2X levels using 6-31+g(d) basis sets (LANL2DZ basis sets for metal atom) in this work. The calculated βHRS(−2ω; ω, ω) values are compared in Fig. 3. It can be found that a substantial enhancement of the second-order NLO response has been obtained in one-electron-oxidized process, while the second-order NLO response has no change after two-electron oxidization. As mentioned above, the one-electron-oxidized center of H is TTF unit. This indicates that one-electron oxidization will affect the geometrical structure of the TTF unit. The crystallographic study84 and our DFT calculations both show that the TTF unit in neutral H forms a bent structure, while the bent TTF unit changes to a planar structure after one-electron oxidization. Due to the two-electron-oxidized center is the Fc unit, the TTF unit is still planar after two-electron oxidization. This result shows that the first hyperpolarizability is sensitive to the structural transformation of the TTF unit in the series of oxidized processes. The planar TTF unit with the well conjugated feature will enhance the second-order NLO response.
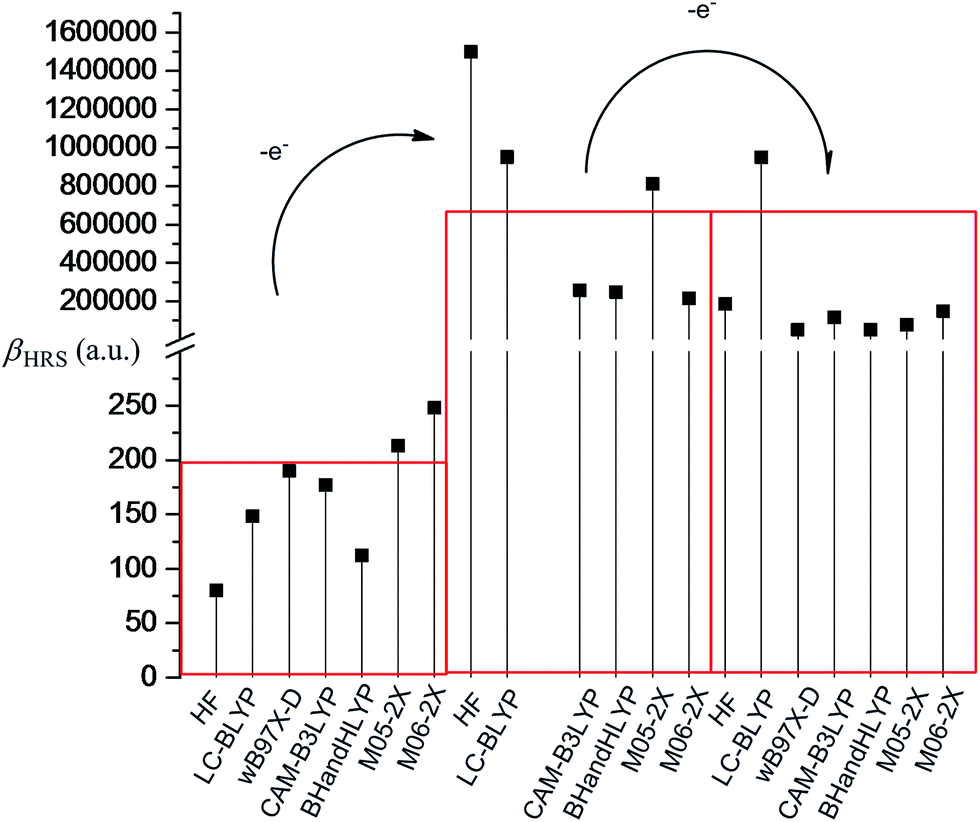 | ||
| Fig. 3 Comparisons of the frequency-dependent first hyperpolarizability of H in different redox states (The wB97X-D-derived βHRS(−2ω; ω, ω) value of the one-electron-oxidized species has not been achieved because of the bad SCF convergence). | ||
The TDDFT calculated excited energy and oscillator strength of the crucial excited state for H, 2H+oxidized and antiH2+oxidized has been listed in Table 3. It can be found that calculated oscillator strength of the three species is on the same order. The one-electron-oxidized process leads to a 4-fold decrease of the excited energy, where it decreases from 3.96 to 0.96 eV. By contrast, the two-electron-oxidized process does not largely affect the magnitude of the excited energy. The calculated excited energy changes from 0.96 to 1.29 eV. According to the two-level model, the decrease of excited energy would enhance the second-order NLO properties, which is in agreement with the hyperpolarizability calculation.
| Compounds | fos | ΔE (eV) | Major contributions |
|---|---|---|---|
| H | 0.21 | 3.96 | HOMO → LUMO + 6 (+41%) |
| HOMO → LUMO + 9 (+22%) | |||
| 2H+oxidized | 0.34 | 0.96 | βHOMO → βLUMO (100%) |
| antiH2+oxidied | 0.26 | 1.29 | HOMO → LUMO (100%) |
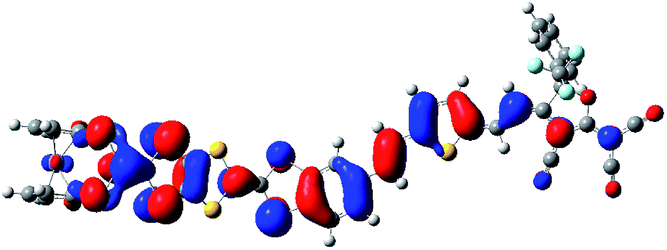
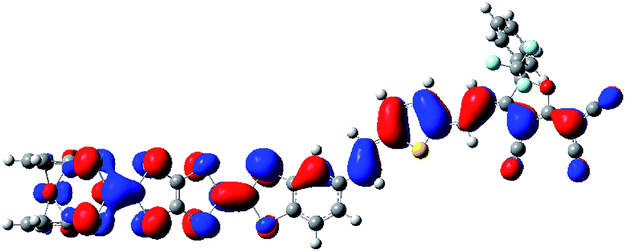
In the purpose of improvement of the role of Fc unit in switching of second-order NLO response in the series of oxidized process, the system 2 may be a well candidate because the Fc unit acts as the role of a bridge between TTF unit and π-A moiety (see Fig. 1). It is well known that the redox property of molecule is closely associated with the nature of frontier molecular orbital. As shown in Fig. 4, the HOMO of 2 is mainly localized on the TTF unit. This indicates that the one-electron-oxidized center of 2 may be the TTF unit. Thus, unrestricted optimized calculations of one-electron-oxidized species 22+oxidized (the one-electronic-oxidized species has one unpaired electron, and thus a doublet state), are performed to check predictions made by molecular orbital analysis. It can be seen from Fig. 4 that the spin density of 22+oxidized mainly localizes on the exTTF unit, which is well in agreement with the molecular orbital predictions. For two-electron-oxidized species of 22+oxidized, two possible spin states (singlet and triplet state, 122+oxidized and 322+oxidized) have been optimized at B3LYP/6-31g(d) level (LANL2DZ basis set on metal ion). The results indicate that the singlet state is 40.33 kJ mol−1 lower than the triplet state. The NBO charges of Fe(II) atom and TTF unit for 2, 22+oxidized and 122+oxidized have been calculated at B3LYP levels using 6-31g(d) basis sets (LANL2DZ basis set on metal ion), and have been listed in Table 4. It can be found that the calculated NBO charge of the TTF unit is positive and Fe(II) center is negative. The NBO charge of TTF unit gets a significantly increase in one- and two-electron-oxidized processes (0.18 for 2, 0.49 for 22+oxidized, and 1.54 for 122+oxidized). By contrast, the NBO charge of the Fe(II) center does not change largely in the series of oxidized processes. This result also supports that TTF unit is the oxidized center in the series of oxidized processes.
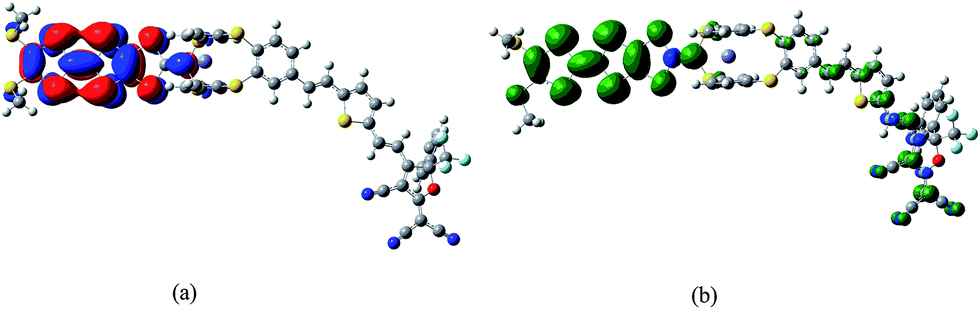 | ||
| Fig. 4 The HOMO of 2 (a) and the spin density distribution of its one-electron-oxidized species 22+oxidized (b). | ||
| Compounds | Functionals | βHRS | DR | Q(TTF) | Q(Fe) | fos | ΔE (eV) |
|---|---|---|---|---|---|---|---|
| a 1 a.u. = 3.62 × 10−42 m4 V−1 = 8.64 × 10−33 esu. | |||||||
| 2 | LC-BLYP | 69202 |
4.98 | 0.18 | −0.26 | 1.33 | 2.26 |
| CAM-B3LYP | 208214 |
5.00 | |||||
| M05-2X | 201306 |
5.00 | |||||
| M06-2X | 229712 |
5.00 | |||||
| 22+oxidized | LC-BLYP | 595922 |
5.15 | 0.49 | −0.13 | 1.46 | 2.24 |
| CAM-B3LYP | 71464 |
3.43 | |||||
| M05-2X | 266744 |
1.59 | |||||
| M06-2X | 71541 |
3.95 | |||||
| 122+oxidized | LC-BLYP | 142565526 |
2.06 | 1.54 | −0.28 | 0.18 | 0.41 |
| CAM-B3LYP | 924110 |
4.00 | |||||
| M05-2X | 2888725 |
2.15 | |||||
| M06-2X | 13657080 |
3.68 | |||||
The frequency-dependent hyperpolarizability of 22+oxidized and 122+oxidized has been calculated at LC-BLYP, CAM-B3LYP, M05-2X, and M06-2X levels using 6-31+g(d) basis sets (LANL2DZ basis sets for metal atom) in this work. The calculated βHRS(−2ω; ω, ω) values have been listed in Table 4. It can be found that the LC-BLYP functional gives the largest βHRS(−2ω; ω, ω) values for the one- and two-electron-oxidized species and the smallest βHRS(−2ω; ω, ω) value for the neutral system 2 among all four functionals, and thus the LC-BLYP functional overestimates the redox-switchable effects on optical nonlinearity when compared with the CAM-B3LYP-, M05-2X-, and M05-2X-derived results. All four functionals show that the two-electron-oxidized process significantly enhances the second-order NLO property. Although the one-electron-oxidized process also leads to an enhancement of the βHRS(−2ω; ω, ω) value, this enhancement is weak relevant to the two-electron-oxidized process according to our DFT calculations. On the other hand, the DR values of 22+oxidized and 122+oxidized are smaller than that of 2, which indicates that the NLO-phore is off ideal one-dimensional structures.
The TDDFT calculated excited energy and oscillator strength of the crucial excited state for 2, 22+oxidized and 122+oxidized also has been listed in Table 4. The results indicate that the excited energy and oscillator strength are not sensitive to the one-electron-oxidized process, and are on the same order of magnitude for 2 and 22+oxidized. It is well-known that the first hyperpolarizability is inversely proportional to the cube of excited energy and proportional to the oscillator strength, the analogous magnitudes of excited energy and oscillator strength imply small change of the first hyperpolarizability (see Table 4). Thus it will lead to a small enhancement on the first hyperpolarizability in the one-electron-oxidized process. By contrast, the crucial excited energies of 122+oxidized are significantly lower than that of 2 and 22+oxidized, the crucial excited energies of 122+oxidized are ∼5.6 and ∼5.3 times as small as that of 2 and 22+oxidized, respectively. The very low excited energy will generate a substantial increase in the second-order NLO responses, which is well in agreement with the hyperpolarizability calculations with all four functionals.
4. Conclusions
In the present paper, the HF and various DFT XC functionals have been employed to evaluate the HRS responses of a series of Fc–TTF hybrids. The total HRS first hyperpolarizability, βHRS(−2ω; ω, ω), has been calculated in gas phase within the T convention. The 6-31+g(d) basis sets containing diffuse and polarization functions have been used. The calculated HRS hyperpolarizabilities are dependent on the XC functionals: the long-range corrected LC-BLYP and wB97X-D functionals provide good performances on the computed βHRS(−2ω; ω, ω) values of the series of Fc–TTF hybrids.The donor strength of the Fc–TTF unit in the D-π-A structure has been elucidated through the HRS hyperpolarizability and TDDFT calculations. We found that the calculated hyperpolarizability is not sensitive to the presence of Fc unit. And the electron density distributions on the Fc unit in the series of frontier molecular orbitals (HOMO, HOMO−1, and HOMO−2), which is associated with the crucial excited state, is nearly zero. All results indicate that Fc unit in the Fc–TTF hybrid does not display the role of electron donor. Duo to the Fc–TTF hybrid possesses a series of reversible oxidation waves, the redox-switchable second-order NLO responses of Fc–TTF hybrid have been explored based on our DFT calculations. The HRS hyperpolarizabilities here computed for the one- and two-electron-oxidization have been compared. The calculated HRS hyperpolarizability are not affected in any significant way by the two-electron-oxidization. Differently, when going from the neutral Fc–TTF hybrid to the one-electron-oxidized species, the HRS hyperpolarizability remarkably increases. Interestingly, in another system the two-electron-oxidization significantly enhances the HRS hyperpolarizability, while HRS hyperpolarizability is not largely affected by the one-electron-oxidization.
Acknowledgements
The authors gratefully acknowledge the financial support from the National Natural Science Foundation of China (21373043), Chinese Postdoctoral Science Foundation (2013M540261), and the Scientific Research Foundation for Doctor of Northeast Dianli University (BSJXM-201110).References
- M. Albota, D. Beljonne, J.-L. Brédas, J. E. Ehrlich, J.-Y. Fu, A. A. Heikal, S. E. Hess, T. Kogej, M. D. Levin, S. R. Marder, D. McCored-Maughon, J. Perry, W. H. Rockel, M. Rumi, G. Subramaniam, W. W. Webb, X.-L. Wu and C. Xu, Science, 1998, 281, 1653–1656 CrossRef CAS.
- Y. Shi, C. Zhang, H. Zhang, J. H. Bechtel, L. R. Dalton, B. H. Robinson and W. H. Steier, Science, 2000, 288, 119–122 CrossRef CAS.
- M. Lee, H. E. Katz, C. Erben, D. M. Gill, P. Gopalan, J. D. Heber and D. J. McGee, Science, 2002, 298, 1401–1403 CrossRef CAS PubMed.
- B. J. Coe, S. Houbrechts and I. Asselberghs, Angew. Chem., Int. Ed., 1999, 38, 366–369 CrossRef CAS.
- B. J. Coe, Acc. Chem. Res., 2006, 39, 383–393 CrossRef CAS PubMed.
- C. Sporer, I. Ratera, D. Ruiz-Molina, Y. Zhao, J. Vidal-Gancedo, K. Wurst, P. Jaitner, K. Clays, A. Persoons, C. Rovira and J. A. Veciana, Angew. Chem., Int. Ed., 2004, 43, 5266–5268 CrossRef CAS PubMed.
- M. Malaun, Z. R. Reeves, R. L. Paul, J. C. Jeffery, J. A. McCleverty, M. D. Ward, I. Asselberghs, K. Clays and A. Persoons, Chem. Commun., 2001, 49–50 RSC.
- K. A. Green, M. P. Cifuentes, M. Samoc and M. G. Humphrey, Coord. Chem. Rev., 2011, 255, 2530–2541 CrossRef CAS PubMed.
- B. J. Coe, Coord. Chem. Rev., 2013, 257, 1438–1458 CrossRef CAS PubMed.
- S. Muhammad, H. L. Xu, M. R. S. A. Janjua, Z. M. Su and M. Nadeem, Phys. Chem. Chem. Phys., 2010, 12, 4791–4799 RSC.
- F. Mancuois, L. Sanguinet, J.-L. Pozzo, M. Guillaume, B. Champagne, V. Rodriguez, F. Adamietz, L. Ducasse and F. Castet, J. Phys. Chem. B, 2007, 111, 9795–9802 CrossRef PubMed.
- L. Sanguinet, J.-L. Pozzo, V. Rodriguez, F. Adamietz, F. Castet, L. Ducasse and B. Champagne, J. Phys. Chem. B, 2005, 109, 11139–11150 CrossRef CAS PubMed.
- C. G. Liu, Z. M. Su, X. H. Guan and S. Muhammad, J. Phys. Chem. C, 2011, 115, 23946–23954 CAS.
- C. G. Liu and X. H. Guan, Phys. Chem. Chem. Phys., 2012, 14, 5297–5306 RSC.
- S. Muhammad, M. R. S. A. Janjua and Z. M. Su, J. Phys. Chem. C, 2009, 113, 12551–12557 CAS.
- S. Muhammad, T. Minani, H. Fukui, K. Yoneda, R. Kishi, Y. Shigeta and M. Nakano, J. Phys. Chem. A, 2011, 116, 1417–1424 CrossRef PubMed.
- S. Muhammad, C. G. Liu, L. Zhao, S. X. Wu and Z. M. Su, Theor. Chem. Acc., 2009, 122, 77–86 CrossRef CAS.
- J. Becher, J. O. Jeppesen and K. Nielsen, Synth. Met., 2003, 133–134, 309–315 CrossRef CAS.
- J. O. Jeppesen and J. Becher, Eur. J. Org. Chem., 2003, 2003, 3245–3266 CrossRef PubMed.
- M. R. Bryce, J. Mater. Chem., 2000, 10, 589–598 RSC.
- J. L. Segura and N. Martín, Angew. Chem., Int. Ed., 2001, 40, 1372–1409 CrossRef CAS.
- D. Canevet, M. Sallé, G. Zhang, D. Zhang and D. Zhu, Chem. Commun., 2009, 2245–2269 RSC.
- M. B. Nielsen, C. Lomholt and J. Becher, Chem. Soc. Rev., 2000, 29, 153–164 RSC.
- J. F. Lamère, I. Malfant, A. Sournia-Saquet and P. G. Lacroix, Chem. Mater., 2007, 19, 805–815 CrossRef.
- C. G. Liu, W. Guan, P. Song, L. K. Yan and Z. M. Su, Inorg. Chem., 2009, 48, 6548–6554 CrossRef CAS PubMed.
- C. G. Liu, X. H. Guan and Z. M. Su, J. Phys. Chem. C, 2011, 115, 6024–6032 CAS.
- C. G. Liu, Mol. Phys., 2014, 112, 199–205 CrossRef CAS.
- G. Guirado, C. Coudret and J.-P. Jaunay, J. Phys. Chem. C, 2007, 111, 2770–2776 CAS.
- B. J. Coe, R. J. Docherty, S. P. Foxon, E. C. Harper, M. Helliwell, J. Raftery, K. Clays, E. Franz and B. S. Brunschwig, Organometallics, 2009, 28, 6880–6892 CrossRef CAS.
- Y. Liao, B. E. Eichinger, K. A. Firestone, M. Haller, J. Luo, W. Kaminsky, J. B. Benedict, P. J. Reid, A. K.-Y. Jen, L. R. Dalton and B. H. Robinson, J. Am. Chem. Soc., 2005, 127, 2758–2766 CrossRef CAS PubMed.
- A. Trujillo, M. Fuentealba, D. Carrillo, C. Manzur, I. Ledoux-Pak, J.-R. Hamon and J.-Y. Saillard, Inorg. Chem., 2010, 49, 2750–2764 CrossRef CAS PubMed.
- G. Li, Y. Song, H. Hou, L. Li, Y. Fan, Y. Zhu, X. Meng and L. Mi, Inorg. Chem., 2003, 42, 913–920 CrossRef CAS PubMed.
- T. L. Kinnibrugh, S. Salman, Y. A. Getmanenko, V. Coropceanu III, W. W. Porter, T. V. Timofeeva, A. J. Matzger, J.-L. Brédas, S. R. Marder and S. Barlow, Organometallics, 2009, 28, 1350–1357 CrossRef CAS PubMed.
- B. J. Coe, J. Fielder, S. P. Foxon, I. Asselberghs, K. Clays and B. S. Van Cleuvenbergen, Organometallics, 2011, 30, 5731–5743 CrossRef CAS.
- H. K. Sharma, K. H. Pannell, I. Ledoux, J. Zyss, A. Ceccanti and P. Zanello, Organometallics, 2000, 19, 770–774 CrossRef CAS.
- S. D. Bella, Chem. Soc. Rev., 2001, 30, 355–366 RSC.
- M. Malaun, Z. R. Reeves, R. L. Paul, J. C. Jeffery, J. A. McCleverty, M. D. Ward, I. Asselberghs and K. A. Clays, Chem. Commun., 2001, 49–50 RSC.
- A. J. Moore, P. J. Skabara, M. R. Bryce, A. S. Batsanov, J. A. K. Howard and S. T. A. K. Daley, J. Chem. Soc., Chem. Commun., 1993, 4, 417–419 RSC.
- S. B. Wilkes, I. R. Butler, A. E. Underhill, A. Kobayashi and H. Kobayashi, J. Chem. Soc., Chem. Commun., 1994, 1, 53–54 RSC.
- S. B. Wilkes, I. R. Butler, A. E. Underhill, M. B. Hursthouse, D. E. Hibbs and K. M. A. Malik, J. Chem. Soc., Dalton Trans., 1995, 7, 897–903 RSC.
- (a) H.-J. Lee, D.-Y. Noh, A. E. Underhill and C.-S. Lee, J. Mater. Chem., 1999, 9, 2359–2363 RSC; (b) P. J. Skabara, M. R. Bryce, A. S. Batsanov and J. A. K. Howard, J. Org. Chem., 1995, 60, 4644–4646 CrossRef CAS.
- R. Andreu, J. Garin, J. Orduna, M. Saviron and S. Uriel, Synth. Met., 1995, 70, 1113–1114 CrossRef CAS.
- H.-J. Lee and D.-Y. Noh, J. Mater. Chem., 2000, 10, 2167–2172 RSC.
- M. Iyoda, T. Takano, N. Otani, K. Ugawa, M. Yoshida, H. Matsuyama and Y. Kuwatani, Chem. Lett., 2001, 30, 1310–1311 CrossRef.
- U. T. Mueller-Westerhoff and R. W. Sanders, Organometallics, 2003, 22, 4778–4782 CrossRef CAS.
- T. Kusamoto, K. Takada, R. Sakamoto, S. Kume and H. Nishihara, Inorg. Chem., 2012, 51, 12102–12113 CrossRef CAS PubMed.
- K. A. Green, M. P. Cifuentes, T. C. Corkery, M. Samoc and M. G. Humphrey, Angew. Chem., Int. Ed., 2009, 48, 7867–7870 CrossRef CAS PubMed.
- K. Szacilowski, Chem. Rev., 2008, 108, 3481–3548 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford CT, 2009.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200–206 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–284 CrossRef CAS PubMed.
- W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284–299 CrossRef CAS PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–311 CrossRef CAS PubMed.
- J. Olsen and P. Jorgensen, J. Chem. Phys., 1985, 82, 3235–3264 CrossRef CAS PubMed.
- H. Sekino and R. J. Bartlett, J. Chem. Phys., 1986, 85, 976–989 CrossRef CAS PubMed.
- J. E. Rice and N. C. Handy, J. Chem. Phys., 1991, 94, 4959–4971 CrossRef CAS PubMed.
- J. E. Rice, R. D. Amos, S. M. Colwell, N. C. Handy and J. Sanz, J. Chem. Phys., 1990, 93, 8828–8839 CrossRef CAS PubMed.
- J. E. Rice and N. C. Handy, Int. J. Quantum Chem., 1992, 43, 91–118 CrossRef CAS PubMed.
- F. Castet and B. Champagne, J. Chem. Theory Comput., 2012, 8, 2044–2052 CrossRef CAS.
- H. Iikura, T. Tsuneda, T. Yanai and K. Hirao, J. Chem. Phys., 2001, 115, 3540–3544 CrossRef CAS PubMed.
- Y. Zhao, N. E. Schultz and D. G. Truhlar, J. Chem. Phys., 2005, 123, 1611031–1611034 Search PubMed.
- Y. Zhao, N. E. Schultz and D. G. Truhlar, J. Chem. Theory Comput., 2006, 2, 364–382 CrossRef.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS PubMed.
- J.-D. Chai and M. Head-Gordon, J. Chem. Phys., 2008, 128, 084106 CrossRef PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- T. Yanai, D. Tew and N. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS PubMed.
- M. Guillaume, B. Champagne, N. Markova, V. Enchev and F. Castet, J. Phys. Chem. A, 2007, 111, 9914–9923 CrossRef CAS PubMed.
- F. Mançois, L. Sanguinet, J.-L. Pozzo, M. Guillaume, B. Champagne, V. Rodriguez, F. Adamietz, L. Ducasse and F. Castet, Acido-Triggered Nonlinear Optical Switches: Benzazolo-oxazolidines, J. Phys. Chem. B, 2007, 111, 9795–9802 CrossRef PubMed.
- A. Plaquet, M. Guillaume, B. Champagne, L. Rougier, F. Mançois, V. Rodriguez, J.-L. Pozzo, L. Ducasse and F. I. Castet, J. Phys. Chem. C, 2008, 112, 5638–5645 CAS.
- A. Plaquet, M. Guillaume, B. Champagne, F. Castet, L. Ducasse, J.-L. Pozzob and V. Rodriguez, Phys. Chem. Chem. Phys., 2008, 10, 6223–6232 RSC.
- F. Mançois, J.-L. Pozzo, J. Pan, F. Adamietz, V. Rodriguez, L. Ducasse, F. Castet, A. Plaquet and B. Champagne, Chem.–Eur. J., 2009, 15, 2560–2571 CrossRef PubMed.
- E. Bogdan, L. Rougier, L. Ducasse, B. Champagne and F. Castet, J. Phys. Chem., 2010, 114, 8474–8479 CrossRef CAS PubMed.
- E. Bogdan, A. Plaquet, L. Antonov, V. Rodriguez, L. Ducasse, B. Champagne and F. Castet, J. Phys. Chem., 2010, 114, 12760–12768 CAS.
- B. Champagne, A. Plaquet, J.-L. Pozzo, V. Rodriguez and F. Castet, J. Am. Chem. Soc., 2012, 134, 8101–8103 CrossRef CAS PubMed.
- R. Bersohn, Y. H. Pao and H. L. Frisch, J. Chem. Phys., 1966, 45, 3184–3199 CrossRef CAS PubMed.
- F. Castet, E. Bogdan, A. Plaquet, L. Ducasse, B. Champagne and V. Rodriguez, J. Chem. Phys., 2012, 136, 024506–024521 CrossRef PubMed.
- D. Qi, NLO Calculator, version 0.2, University of Science and Technology Beijing, Beijing, China, 2012 Search PubMed.
- L. J. Zhang, D. D. Qi, L. Y. Zhao, C. Chen, Y. Z. Bian and W. J. Li, J. Phys. Chem., 2012, 116, 10249–10256 CrossRef CAS PubMed.
- A. Willetts, J. E. Rice, D. M. Burland and D. P. Shelton, J. Chem. Phys., 1992, 97, 7591–7599 Search PubMed.
- M. A. L. Marques and A. Rubio, Phys. Chem. Chem. Phys., 2009, 11, 4436 RSC.
- T. Kusamoto, H. Nishihara and R. Kato, Inorg. Chem., 2013, 52, 13809–13811 CrossRef CAS PubMed.
- L. R. Dalton, P. A. Sullivan and D. H. Bale, Chem. Rev., 2010, 110, 25–55 CrossRef CAS PubMed.
- Y. J. Cheng, J. D. Luo, S. Hau, D. H. Bale, T.-D. Kim, Z. Shi, D. B. Lao, N. M. Tucker, Y. Tian, L. R. Dalton, P. J. Reid and A. K. Y. Jen, Chem. Mater., 2007, 19, 1154–1163 CrossRef CAS.
- J. L. Oudar and D. S. Chemla, J. Chem. Phys., 1977, 66, 2664–2668 CrossRef CAS PubMed.
- J. L. Oudar, J. Chem. Phys., 1977, 67, 446–457 CrossRef CAS PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem., 2009, 113, 6378–6396 CrossRef CAS PubMed.
- A. Lewis, J. A. Bumpus, D. G. Truhlar and C. J. Cramer, J. Chem. Educ., 2004, 81, 596–604 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: xyz coordinates for all structures reported in this paper. See DOI: 10.1039/c4ra04548c |
| ‡ Present Addresses: No. 5625, Renmin Road, Changchun City, P. R. China. |
| This journal is © The Royal Society of Chemistry 2014 |