Highly porous carbons with superior performance for CO2 capture through hydrogen-bonding interactions
Donghai Lina,
Xiaotian Zhanga,
Xinwei Cuib and
Weixing Chen*a
aDepartment of Chemical and Materials Engineering, University of Alberta, Edmonton, Alberta, Canada T6G 2G6. E-mail: weixing.chen@ualberta.ca
bAdvEn Solutions Inc., 3231 Trdger Close, Edmonton, Alberta, Canada T6R 3T6
First published on 9th June 2014
Abstract
Highly porous carbons were prepared by using polyaniline (PANI) as a carbon precursor and KOH as an activating agent via a one-step chemical activation process. The effects of the activation parameters such as activation temperature, KOH–PANI weight ratio and pre-heating temperature were fully investigated, through which the pore structure and the materials chemistry of the activated porous carbons were optimized. When studied as an adsorbent for CO2 capture, the optimized porous carbon exhibited a high CO2 capture capacity of 4.50 mmol g−1, high multi-cycle sorption/desorption stability and highly selective adsorption of CO2 over N2 (0.27 mmol g−1) at 25 °C. This superior performance for CO2 capture was found to be closely related to C–H groups on the carbon surface through hydrogen bonding interactions.
1. Introduction
Reduction of CO2 concentration in the atmosphere has attracted considerable attention because CO2 is considered to be the main anthropogenic contributor to climate change.1–3 In addition, a high concentration of CO2 is harmful to humans, especially in space-limited chambers like submarines and space ships.4 The selective capture and storage of CO2 at low cost in an energy-effective way are crucial for achieving substantial reduction of the emission level.3,5–7Most of the current commercial technologies for CO2 capture are based on absorption–regeneration technology involving liquid amine-based process.8,9 However, this technology has the problems of high energy consumption, solvent degradation, and equipment corrosion.10,11 In order to overcome the disadvantages of the liquid amine-based process, researchers focused on the adsorption by solid adsorbents because of its low energy consumption and low equipment cost. Up to now, a large number of solid adsorbents have been employed for CO2 capture, such as zeolites,12–15 mesoporous silica,5,16–18 metal–organic frameworks (MOFs),19,20 metal oxides,21 ionic liquid22 and porous carbons.23–28 Carbon-based adsorbents are most promising for CO2 capture due to high chemical stability, low cost, high surface area and large adsorption capacity.7,29,30
High surface area and/or high pore volume are the prerequisites for CO2 capture by CO2 gas physisorption27,31 In addition, CO2 is slightly acidic, and thus the basicity of adsorbents in surface chemistry plays an important role in achieving high CO2 capture performance through acid–base interactions.32 Adsorbents functionalized with amines show high CO2 adsorption capacity.33 When nitrogen is incorporated or doped to porous carbons, their CO2 adsorption capacity will be further improved.25,34,35 However, the influence of N-containing groups on CO2 capture is still controversial and not well-defined.31 One-to-one base–acid interaction cannot explain the fact that, in some cases, a small amount of doped nitrogen results in greatly increased CO2 adsorption capacity.
Conducting polymer, polyaniline (PANI), has been intensively studied due to its facile synthesis, low cost, environmental stability and unique optical, electrical, and electrochemical properties.36 This polymer has been previously employed as a precursor to fabricate N-doped porous carbons by means of nanocasting techniques. Two-step chemical activation process, carburization and then activation, was normally adopted to produce porous carbons using conducting polymers as carbon precursors.6,23,25,37–39 However, to our best knowledge, no work has been reported to produce N-doped, highly porous carbons from PANI through a one-step chemical activation process.
In this paper, PANI was used as the carbon precursor and KOH as the activating agent to produce N-doped, highly porous carbons through a one-step chemical activation process. The effects of the activation parameters such as activation temperature, KOH–PANI weight ratio and pre-heating temperature were fully investigated and optimized. The resulted activated carbon exhibited a high CO2 capture capacity of 4.50 mmol g−1, high multi-cycle sorption/desorption stability and highly selective adsorption of CO2 over N2 (0.27 mmol g−1) at 25 °C. The contributions to the CO2 capture performance from the specific surface area and N content were discussed. It was found that, in addition to these two factors, the superior performance for CO2 capture is closely related to C–H groups on the carbon surface through hydrogen bonding interactions.
2. Experimental
2.1 Synthesis of highly porous carbons
PANI was prepared by aniline oxidation using ammonium persulfate as an oxidant. Briefly, aniline (0.23 M, 350 mL) acid solution was slowly dropped into an ammonium persulfate (0.54 M, 150 mL) acid solution at ice water temperature and the mixture was magnetically stirred for 6 h. Dark green PANI was then separated by filtration and washed thoroughly with de-ionized water until neutral pH was reached and then dried. The final PANI yield was around 100%. A physical mixture of KOH and PANI (weight ratio of KOH–PANI = 1–6) was preheated at (150–400 °C, 1 h) in a tube furnace under Ar flow (100 mL min−1), followed by a high-temperature (500–700 °C, 1 h) activation process. The adsorbents were obtained by consequent washing the residue with de-ionized water and ethanol until neutral pH was reached and then dried in an oven at 120 °C. The as-synthesized samples were denoted as AC-x-y-z, where x is the KOH–PANI weight ratio, y is the preheating temperature (in °C) and z is the activation temperature (in °C). For example, sample AC-2-200-600 reflects the activation conditions KOH–PANI ratio of 2, preheating at 200 °C for 1 h and activation temperature at 600 °C 1 h. Sample AC-2-600D was prepared by the same method but omitted the preheating process. In order to determine the effect of nitrogen content, we use the sucrose as precursor instead of PANI polymer but the same activation denoted as sucrose-2-200-600. The conventional two-step activation procedure, PANI carbonization at 600 °C, then followed the same preparation process of AC-2-200-600 was identified as AC600-2-200-600.2.2 Material characterization
The morphologies of the samples were examined by scanning electron microscopy (SEM) using a JEOL 6301F. X-ray photoelectron spectroscopy (XPS) was carried out with Kratos Axis 165 using a monochromated Al Kα excitation source. Elemental analyses were done on an Elementar Vario EL III microanalyzer. N2 adsorption–desorption isotherms were measured at 77 K with Autosorb I (Quantachrome, USA). The samples were degassed in a high vacuum for 2 h at 250 °C before nitrogen adsorption measurement. The total pore volume was calculated as the adsorbed volume of liquid nitrogen at a relative pressure of 0.997 and surface area was measured based on the multipoint Brunauer–Emmett–Teller (BET) method at a relative pressure of 0.05–0.20. The pore size distributions (PSDs) and the volume of micropores smaller than a certain pore size were obtained from the N2 adsorption isotherms using the non-local density functional theory (NLDFT) model, assuming a slit pore shape.2.3 CO2 capture measurements
CO2 adsorption capacity was measured using a thermo-gravimetric apparatus (SDT Q600, TA Instruments, USA). The adsorbent particles were ground and sieved to 150–250 μm size range before use. The CO2 capture was tested at TGA (SDT Q600, TA Instruments, USA), the samples were degassed at 105 °C 30 min, then 95% CO2 + 5% N2 (flow rate 100 mL min−1) at 25 °C about 1 h. In the case of adsorption and desorption, during the adsorption, the carbon sample was exposed to a stream of CO2 (100 mL min−1). Once the sample was saturated, the CO2 was switched to He (100 mL min−1) and CO2 was desorbed. The CO2 adsorption experiments were also performed with ASAP 2020C (Micrometrics Corp.) at different temperatures (25, 50, 70, 95 °C). In the case of the adsorption–desorption cycles, the sample (≈30 mg) was degassed under the He gas at 200 °C before the cyclic experiments. During the adsorption, the carbon sample was exposed to a stream of pure CO2 (100 mL min−1). Once the sample was saturated, the gas was switched back from CO2 to He (100 mL min−1) and the carbon dioxide was desorbed. This adsorption–desorption cycle was repeated several times.3. Results and discussion
Scanning electron microscopy (SEM) was used to image the microscopic morphology of the carbon precursor and the activated carbons. PANI, when used as the carbon precursor, exhibited a typical sponge-like architecture formed by interconnected microparticles of about 100 nm in diameter (Fig. 1a). This morphology offers a large interfacial area for the polymer to react with KOH, which is essential to achieve uniform activation reactions and a good pore development. The activated carbons demonstrate a variety of shapes depending on the degree of activation. The less-activated sample AC-1-200-600 (Fig. 1b) contains small pores on the surface, while the fully activated carbon AC-2-200-600 is made up of particles with large cavities and smooth surfaces (Fig. 1c). The over-activated carbon AC-4-200-600 is shown to have extra-large cavities (Fig. 1d). | ||
| Fig. 1 SEM images showing the structural characteristics of the carbon materials: (a) polyaniline (PANI), (b) sample AC-1-200-600, (c) sample AC-2-200-600 and (d) sample AC-4-200-600. | ||
The N2 sorption and pore size distribution (PSD) of the activated carbons, prepared at different activation temperatures, KOH–PANI weight ratios and preheating temperatures, are displayed in Fig. 2. All the activated carbons show an adsorption isotherm of type I in Fig. 2a, c and e, in which, the curves rapidly achieves a high adsorption plateau at very low relative pressure. It indicates that a large content of micropores exists in these samples.23 The PSD graphs in Fig. 2b, d and f also suggest that these activated carbons are made up of mixed micro- and meso-pores,40 which may be beneficial to the fast dynamics of CO2 sorption.28
 | ||
| Fig. 2 Effect of activation temperature (a and b), KOH ratio (c and d) and preheating temperature (e and f) on N2 adsorption isotherms and pore size distributions. | ||
The effect of the activation temperature on the pore structures of the resulted activated carbons are displayed in Fig. 2a and b. The textural parameters of the activated carbons are also concluded in Table 1. The surface area and the total pore volume for the sample AC-2-200-500 are only 184 m2 g−1 and 0.20 cm3 g−1, respectively (Table 1), suggesting that the polymer was not fully carbonized at 500 °C. When increasing the activation temperature to 600 °C, the surface area and the pore volume increased at a staggering rate to 1668 m2 g−1 and 0.35 cm3 g−1, respectively. Further increasing the temperature, the surface area and the pore volume kept increasing up to 2720 m2 g−1 and 0.42 cm3 g−1, respectively. Fig. 2c and d demonstrates the effect of KOH–PANI weight ratio. From the pore structure data in Table 1, we can clearly see that the KOH–PANI ratio of 2 is the optimum ratio. Any deviation from this ratio will decrease both the surface area and the pore volume. Fig. 2e–f illustrates the importance of the preheating temperature on the final pore textures. Although the preheating temperature has been considered as a critical parameter for the activation process, few literatures studied its effect.41 Fig. 2e and f and Table 1 in this report reveal that, increasing the preheating temperature from 150 to 200 °C leads to higher surface area and volume; however, further increasing the preheating temperature to 300 or 400 °C results in lower surface area and pore volume. An optimum preheating temperature of 200 °C was determined. The effect of the preheating temperature will be discussed in detail later in this report.
| Materials | SBET m2 g−1 | Smicrob m2 g−1 | Vpa cm3 g−1 | Vmicroc cm3 g−1 | N% | N/C weight ratio | H/C weight ratio | CO2 uptaked mmol g−1 | Normalize CO2 uptake mmol CO2 per mmol N | Normalized CO2 uptake mmol CO2 per m2 |
|---|---|---|---|---|---|---|---|---|---|---|
| a Total pore volume at p/po ∼ 0.99.b The micropore surface areas.c Volume of micropores with pore diameters less than 1 nm as determined by the NLDFT method.d CO2 capture capacities of the porous carbons at 25 °C and 0.95 atm. N/C and H/C weight ratio were determined by elemental analysis. | ||||||||||
| AC-1-200-600 | 775 | 724 | 0.34 | 0.19 | 8.30 | 0.1523 | 0.03771 | 2.45 | 0.44 | 3.16 |
| AC-1.5-200-600 | 1473 | 1274 | 0.67 | 0.34 | 7.85 | 0.1463 | 0.04163 | 2.95 | 0.50 | 2.00 |
| AC-2-200-600 | 1668 | 1328 | 0.69 | 0.35 | 5.16 | 0.1208 | 0.05140 | 4.30 | 1.17 | 2.58 |
| AC-2.5-200-600 | 898 | 680 | 0.49 | 0.19 | 5.03 | 0.1176 | 0.03282 | 1.64 | 0.90 | 2.90 |
| AC-3-200-600 | 859 | 631 | 0.40 | 0.17 | 4.09 | 0.08114 | 0.02687 | 1.59 | 1.18 | 1.93 |
| AC-4-200-600 | 802 | 503 | 0.50 | 0.14 | 1.84 | 0.03420 | 0.02186 | 1.55 | 0.42 | 1.85 |
| AC-2-150-600 | 1309 | 1094 | 0.56 | 0.29 | 7.18 | 0.1322 | 0.04594 | 3.85 | 0.75 | 2.93 |
| AC-2-300-600 | 586 | 473 | 0.26 | 0.13 | 5.10 | 0.1180 | 0.04005 | 2.69 | 0.54 | 4.59 |
| AC-2-400-600 | 783 | 579 | 0.41 | 0.16 | 3.93 | 0.1177 | 0.03504 | 2.01 | 0.72 | 2.57 |
| AC-2-200-500 | 184 | 107 | 0.20 | 0.028 | 6.63 | 0.1624 | 0.02493 | 2.23 | 0.47 | 12.12 |
| AC-2-200-700 | 2720 | 1441 | 1.68 | 0.42 | 3.20 | 0.04957 | 0.02588 | 2.55 | 1.11 | 0.94 |
| AC600-2-200-600 | 910 | 790 | 0.47 | 0.20 | 5.25 | — | — | 1.95 | 0.13 | 2.14 |
| Sucrose-2-200-600 | 39 | 10 | 0.29 | 0.0028 | — | — | — | 1.60 | — | — |
High surface area and high pore volume are required for CO2 gas physisorption.27,42 Although some CO2 adsorption data reported in Table 1 appear to support this physisorption mechanism, CO2 adsorption data contradictory to the physisorption mechanism also exist. For example, sample AC-2-200-700 and sample AC-1-200-600 are shown to have almost the same CO2 adsorption capacity (2.55 mmol g−1), but they had a very different specific surface areas (2720 m2 g−1 for AC-2-200-700 and 775 m2 g−1 for AC-1-200-600). This indicates that the CO2 uptake cannot be solely rationalized by the physio-characteristics of the activated carbons, such as specific surface area and/or micropore volume.
The chemisorption of CO2 gas has also been reported by many researchers.3,33,39,42,43 It involves in acid–base interactions between N-containing basic functional groups and acidic CO2 gas. However, the capacity of CO2 capture observed in the investigation does not agree well with the chemisorption mechanism. As seen in Table 1, samples AC-2-200-600 and AC-2.5-200-600 contain almost the same nitrogen content, but the former yields much larger CO2 uptake (4.30 mmol g−1) than the latter (1.64 mmol g−1).
More interestingly, as shown in Table 1, sample AC-1-200-600 has larger specific surface area (775 m2 g−1) and higher nitrogen content (8.3%) than those of AC-3-200-600 (a specific surface area of 586 m2 g−1 and a nitrogen content of 5.1%), but yields smaller CO2 uptake (2.45 mmol g−1) than AC-2-200-600 (2.69 mmol g−1). It implies that a combined consideration of chemisorption and physisorption is still inconsistent with the experimental findings and therefore alternative factors must be explored in order to rationalize the high CO2 capture capacity of 4.30 mmol g−1 achieved.
To explore possible alternative factors affecting CO2 uptake, CO2 capture capacities were normalized by the N content that accommodates the effect of acid–base interactions. As shown in Table 1, sample AC-2-200-600 has a larger specific area (1668 m2 g−1) than that of AC-3-200-600 (859 m2 g−1), but they have almost the same CO2 uptake after normalization. Similarly, CO2 capture capacities were normalized by the specific surface area of the samples (Table 1). Although the N content of sample AC-3-200-600 (4.09%) is much higher than that of AC-4-200-600 (1.84%), both have almost the same CO2 uptake when normalized by the specific surface area of the samples. It is obvious that the physical adsorption by surface area and the acid–base interactions by N content together are not able to explain the CO2 adsorption behaviour found in this study. It further indicates that additional factors may also play an important role in CO2 uptake.
The CO2 adsorption capacity and the H/C weight ratio were plotted as a function of the activation temperature at the fixed KOH–PANI ratio of 2 and the preheating temperature of 200 °C in Fig. 3a. As the activation temperature increases, CO2 adsorption capacity exhibits a volcanic change, with a maximum value of 4.30 mmol g−1 obtained at the activation temperature of 600 °C. Interestingly, the changing trend of H/C weight ratio with the activation temperature coincides with that of CO2 adsorption capacity (Fig. 3a). The H/C weight ratio also exhibits a volcanic change with the highest H/C weight ratio observed at 600 °C.
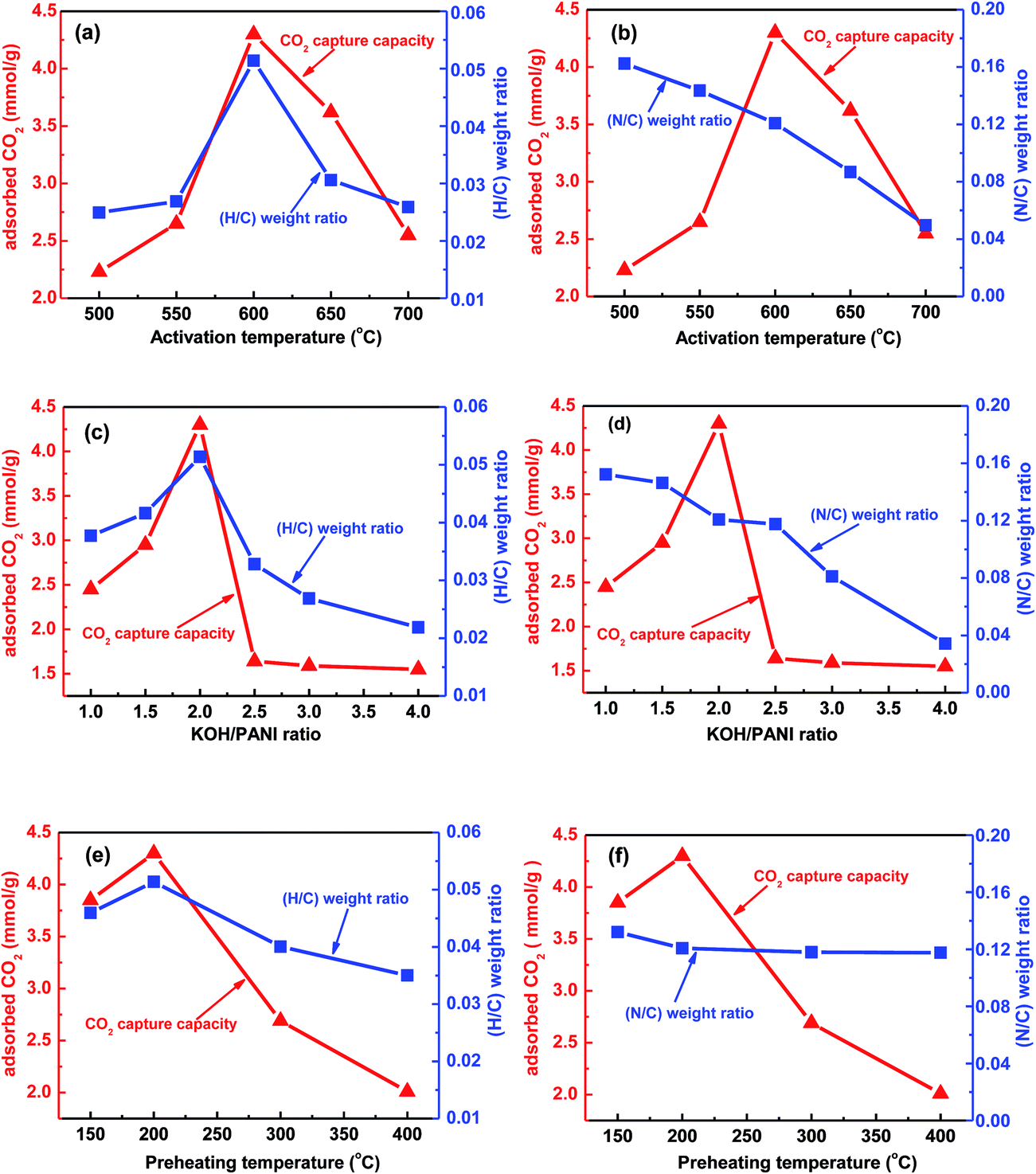 | ||
| Fig. 3 The variation of CO2 adsorption capacities, H/C weight ratio and N/C weight with activation temperature for samples processed with KOH–PANI = 2 and a preheating temperature of 200 °C (a and b), KOH–PANI weight ratio for samples processed at a preheating temperature of 200 °C and an activation temperature of 600 °C (c and d), and preheating temperature for samples processed at KOH–PANI = 2 and an activation temperature of 600 °C (e and f). | ||
In contrast, the N/C weight ratio was found to decrease monotonically with increasing activation temperature (Fig. 3b). Similar trend was also found when the CO2 adsorption capacity, the H/C weight ratio and the N/C weight ratio were plotted as a function of the KOH–PANI ratio in Fig. 3c and d and the preheating temperature in Fig. 3e and f, respectively. In Fig. 3c, the amount of CO2 uptake shows a peak value at a KOH–PANI ratio of 2, which is similar to the variation of the H/C weight ratio with KOH–PANI ratio. However, the N/C weight ratio decreases monotonically from 0.15 to 0.03 with increasing KOH–PANI ratio (Fig. 3d). In Fig. 3e, both the CO2 uptake amount and the H/C weight ratio demonstrate a maximum point at a preheating temperature of 200 °C, while the N/C weight ratio almost keeps the same at a level of around 0.12.
From the above discussion, it is clear that that the trend of CO2 uptake amount is different from that of N/C weight ratio, but correlates well with that of H/C weight ratio. It is, therefore, suggested that the C–H groups on the carbon surface may play a vital role in CO2 capture. The capture of CO2 molecules by C–H group is suggested to be through hydrogen bonding interactions as C–H⋯O. Although the interaction of C–H⋯O is weaker than that of typical hydrogen bonds, the existence of this type of weak hydrogen bond has been validated in the experimental and theoretical studies reported previously.31,44,45 Fig. 4a shows a direct correlation of the CO2 uptake with the H/C weight ratio and a linear relation between the CO2 uptake and the H/C weight ratio is seen. The slope of the linearly fitted line in Fig. 4a was determined to be 143 mmol CO2 g−1 (H/C weight ratio)−1. In contrast, the relation between the CO2 uptake and the N/C weight ratio is statistically insignificant (Fig. 4b). Fig. 4 further confirms the importance C–H group in CO2 capture through hydrogen-bonding interactions. The observation of this type of hydrogen-bonding interaction, C–H⋯O, in CO2 capture may be attributed to the adopted one-step chemical activation process, in which the carburization and the activation occurred concurrently and the formation of C–H groups in the activated carbons were obviously facilitated. The latter was found to contribute to the CO2 capture through hydrogen-bonding interactions.31
 | ||
| Fig. 4 Variation of CO2 adsorption capacities with H/C weight ratio (a) and N/C weight ratio (b) for samples processed at different KOH weight ratios and different preheating temperatures under the activation temperature of 600 °C. | ||
From the above experiment results, it has been determined that the highest CO2 adsorption can be achieved when the KOH–PANI mixtures were activated at the following optimized conditions: the activation temperature of 600 °C for 1 h, the KOH–PANI ratio of 2 and the preheating temperature of 200 °C for 1 h. It is worth to note that, although the pure dry KOH melts at 380 °C, KOH can be melted at a lower temperature when mixed with carbon precursors.43 In this study, we observed that KOH starts to be melted at 200 °C when it was mixed with PANI. It is suggested that, for the preheating temperature lower than 200 °C, the unmelted KOH will not wet the KOH–PANI mixtures thoroughly; for the preheating temperature over 200 °C, the melted KOH may alter the morphology of PANI polymers. In both cases, the pore structures of the final activated carbons will be affected and the CO2 capture capacities will be deteriorated. Therefore, the optimized preheating temperature was obtained at 200 °C.
The CO2 capture behaviour of the porous carbons activated at the optimized conditions was further characterized. As shown in Fig. 5a, the prepared activated carbon AC-2-200-600 has shown a very high CO2 capture capacity of 4.50 mmol g−1 (under 1 atm 25 °C), which is amongst the best of the known porous carbons.35,42,46 The commercial adsorbent SA9T, which is an amine immobilized on the polymer support, indicates only 0.44 mmol g−1 capacity for CO2 uptake.47 Zeolite 13X, which is widely considered to be a promising CO2 adsorbent, presents 3.64 mmol g−1 capacity for CO2 uptake.35 Nitrogen-enriched porous carbons produced from polyacrylonitrile containing block copolymer yields a maximum capacity of 3.00 mmol g−1.46 N-enriched porous carbons by the carbonization of a polymeric material in the presence of an amino acid L-lysine show a maximum CO2 adsorption uptake of 3.1 mmol g−1.34 All these comparison results further reveal the significance of the C–H group on CO2 capture through hydrogen-bonding interactions.
 | ||
| Fig. 5 CO2 adsorption of sample AC-2-200-600 at 25, 50, 70 and 95 °C (a) and isosteric heat of adsorption (Qst) calculated from CO2 adsorption curves (b). | ||
To investigate the CO2 capture performance at elevated temperatures, the adsorption isotherms of AC-2-200-600 at 25, 50, 70 and 95 °C were measured (Fig. 5a). It can be seen that the CO2 uptake decreases significantly with increasing adsorption temperature, which is reasonable since the molecular kinetic energy of CO2 rises with increasing temperature. The higher temperature leads to lower CO2 adsorption capacity, indicating that the CO2 adsorption process on the AC-2-200-600 is an exothermic process. To determine the strength of the interaction between CO2 molecules and the activated AC-2-200-600, the isosteric heat of adsorption (Qst) was calculated by applying Clausius–Clapeyron equation to the CO2 adsorption isotherms at 25, 50, 70 and 95 °C (Fig. 5b).25,34,48 The isosteric heat of adsorption decreases from 36 to 31 kJ mol−1 as the CO2 adsorption amount increases from 0.2 to 1.2 mmol g−1. The high isosteric heat of adsorption is mainly due to hydrogen-bonding interactions (C–H⋯O) and acid–base interactions between N-containing basic functional groups and acidic CO2 gas.
For the scale up application in CO2 sequestration, the adsorbent should also show high stability during cyclic adsorption–desorption processes and high selectivity, in addition to high capacity. The stability during cyclic adsorption–desorption processes for the sample AC-2-200-600 was measured at 25 °C in Fig. 6, which shows only 3% drop in CO2 adsorption capacity after 30 cycles demonstrating its high stability. For selectivity, N2 adsorption on AC-2-200-600 shows a very small adsorption capacity of 0.27 mmol g−1 for N2 gas at 25 °C, indicating that AC-2-200-600 can be used for selective adsorption of CO2 in the atmosphere (Fig. 7a). The selectivity of CO2 over N2 gas, defined as the ratio of the initial slopes of N2 and CO2 adsorption isotherms (Fig. 7b), was calculated using Henry's law, where the initial slopes were calculated from CO2 and N2 adsorption isotherms as reported earlier.47,49 It is interesting that the selectivity of CO2 over N2 was determined to be 36 at 25 °C, which supports the result in Fig. 7a and demonstrates its potential for industrial applications.47 The comparison data in Fig. 5–7 clearly show that the optimized N-doped activated carbons, prepared through a simple and scalable one-step chemical activation process, demonstrate a high adsorption capacity, high stability and high selectivity for CO2 capture.
 | ||
| Fig. 6 Stability of CO2 adsorption–desorption cycles obtained for sample AC-2-200-600 at 25 °C. | ||
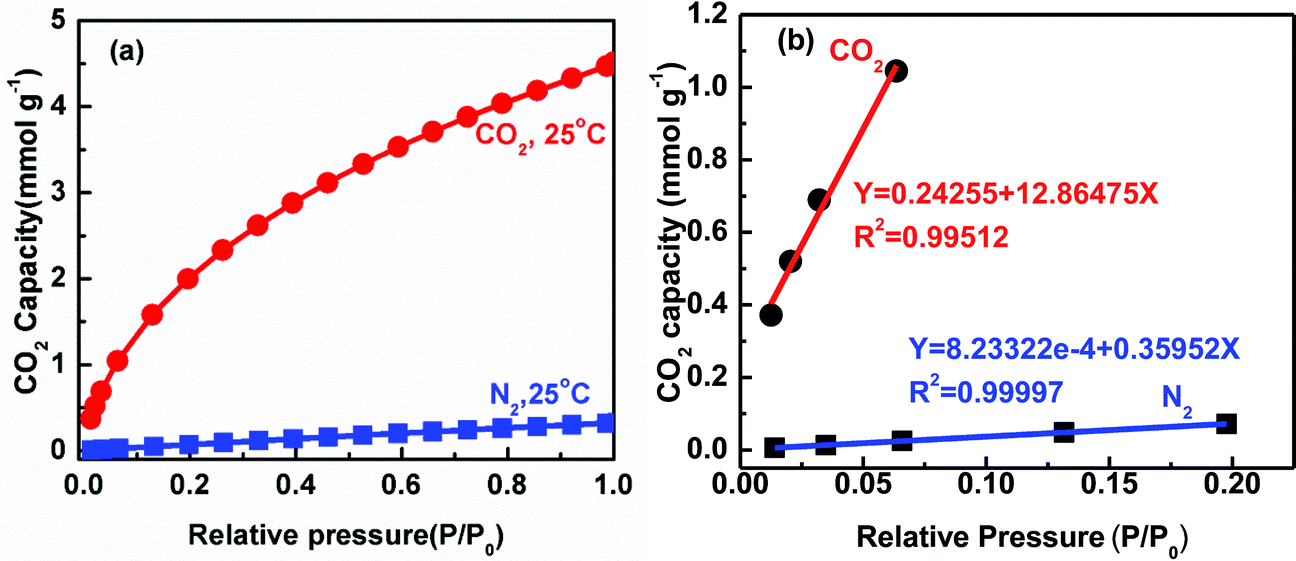 | ||
| Fig. 7 Adsorption curves (a) and determination of initial slopes from CO2 and N2 adsorption isotherms at 25 °C for sample AC-2-200-600 (b). | ||
4. Conclusion
In conclusion, nitrogen-doped, highly porous carbon was synthesized via a one-step chemical activation process by using polyaniline as the carbon precursor and KOH as the activating agent. The effects of the activation parameters such as activation temperature, KOH–PANI weight ratio and pre-heating temperature were fully investigated. The optimum conditions are: the activation temperature of 600 °C for 1 h, the KOH–PANI ratio of 2 and the preheating temperature of 200 °C for 1 h. The activated porous carbon with the optimized pore structure and material's chemistry exhibited a high CO2 capture capacity of 4.50 mmol g−1, high multi-cycle sorption/desorption stability and highly selective adsorption of CO2 over N2 (0.27 mmol g−1) at 25 °C. This superior performance for CO2 capture was found to be closely related to C–H groups on the carbon surface through hydrogen bonding interactions, in addition to specific surface area and N content of the activated carbons. The relatively large content of C–H groups in the activated carbons is believed to be achieved when the carburization process and the activation process occur simultaneously within the one-step chemical activation process.Acknowledgements
The authors would like to acknowledge Carbon Management Canada for the financial support.References
- C. H. Yu, C. H. Huang and C. S. Tan, Aerosol Air Qual. Res., 2012, 12, 745–769 CAS.
- L. Li, N. Zhao, W. Wei and Y. H. Sun, Fuel, 2013, 108, 112–130 CrossRef CAS PubMed.
- S. D. Kenarsari, D. L. Yang, G. D. Jiang, S. J. Zhang, J. J. Wang, A. G. Russell, Q. Wei and M. H. Fan, RSC Adv., 2013, 3, 22739–22773 RSC.
- W. Z. Shen and W. B. Fan, J. Mater. Chem. A, 2013, 1, 999–1013 CAS.
- J. Q. Zhao, F. Simeon, Y. J. Wang, G. S. Luo and T. A. Hatton, RSC Adv., 2012, 2, 6509–6519 RSC.
- G. P. Hao, W. C. Li, D. Qian, G. H. Wang, W. P. Zhang, T. Zhang, A. Q. Wang, F. Schuth, H. J. Bongard and A. H. Lu, J. Am. Chem. Soc., 2011, 133, 11378–11388 CrossRef CAS PubMed.
- A. Samanta, A. Zhao, G. K. H. Shimizu, P. Sarkar and R. Gupta, Ind. Eng. Chem. Res., 2012, 51, 1438–1463 CrossRef CAS.
- R. Bounaceur, N. Lape, D. Roizard, C. Vallieres and E. Favre, Energy, 2006, 31, 2556–2570 CrossRef CAS PubMed.
- E. Favre and H. F. Svendsen, J. Membr. Sci., 2012, 407, 1–7 CrossRef PubMed.
- B. A. Oyenekan and G. T. Rochelle, AIChE J., 2007, 53, 3144–3154 CrossRef CAS.
- W. Yan, J. Tang, Z. J. Bian, J. Hu and H. L. Liu, Ind. Eng. Chem. Res., 2012, 51, 3653–3662 CrossRef CAS.
- J. Kim, L. C. Lin, J. A. Swisher, M. Haranczyk and B. Smit, J. Am. Chem. Soc., 2012, 134, 18940–18943 CrossRef CAS PubMed.
- J. Zhang, R. Singh and P. A. Webley, Microporous Mesoporous Mater., 2008, 111, 478–487 CrossRef CAS PubMed.
- J. Merel, M. Clausse and F. Meunier, Ind. Eng. Chem. Res., 2008, 47, 209–215 CrossRef CAS.
- J. Zhou, W. Li, Z. S. Zhang, W. Xing and S. P. Zhuo, RSC Adv., 2012, 2, 161–167 RSC.
- R. Vaidhyanathan, S. S. Iremonger, G. K. H. Shimizu, P. G. Boyd, S. Alavi and T. K. Woo, Science, 2010, 330, 650–653 CrossRef CAS PubMed.
- D. H. Lin, Y. X. Jiang, Y. Wang and S. G. Sun, J. Nanomater., 2008, 473791, DOI:10.1155/2008/473791.
- N. Hedin, L. J. Chen and A. Laaksonen, Nanoscale, 2010, 2, 1819–1841 RSC.
- L. Ge, L. Wang, V. Rudolph and Z. H. Zhu, RSC Adv., 2013, 3, 25360–25366 RSC.
- P. Nugent, Y. Belmabkhout, S. D. Burd, A. J. Cairns, R. Luebke, K. Forrest, T. Pham, S. Q. Ma, B. Space, L. Wojtas, M. Eddaoudi and M. J. Zaworotko, Nature, 2013, 495, 80–84 CrossRef CAS PubMed.
- F. J. Song, Y. X. Zhao, Y. Cao, J. Ding, Y. F. Bu and Q. Zhong, Appl. Surf. Sci., 2013, 268, 124–128 CrossRef CAS PubMed.
- X. X. Lei, Y. J. Xu, L. L. Zhu and X. H. Wang, RSC Adv., 2014, 4, 7052–7067 RSC.
- M. Sevilla, C. Falco, M. M. Titirici and A. B. Fuertes, RSC Adv., 2012, 2, 12792–12797 RSC.
- K. V. Kumar and F. Rodriguez-Reinoso, RSC Adv., 2012, 2, 9671–9678 RSC.
- Z. S. Zhang, J. Zhou, W. Xing, Q. Z. Xue, Z. F. Yan, S. P. Zhuo and S. Z. Qiao, Phys. Chem. Chem. Phys., 2013, 15, 2523–2529 RSC.
- Z. H. Tang, Z. Han, G. Z. Yang, B. Zhao, S. L. Shen and J. H. Yang, New Carbon Mater., 2013, 28, 55–60 CrossRef CAS.
- M. Zhong, E. K. Kim, J. P. McGann, S. E. Chun, J. F. Whitacre, M. Jaroniec, K. Matyjaszewski and T. Kowalewski, J. Am. Chem. Soc., 2012, 134, 14846–14857 CrossRef CAS PubMed.
- X. Y. Ma, M. H. Cao and C. W. Hu, J. Mater. Chem. A, 2013, 1, 913–918 CAS.
- X. Hu, M. Radosz, K. A. Cychosz and M. Thommes, Environ. Sci. Technol., 2011, 45, 7068–7074 CrossRef CAS PubMed.
- Q. R. Fang, G. S. Zhu, M. Xue, J. Y. Sun, Y. Wei, S. L. Qiu and R. R. Xu, Angew. Chem., Int. Ed., 2005, 44, 3845–3848 CrossRef CAS PubMed.
- W. Xing, C. Liu, Z. Y. Zhou, L. Zhang, J. Zhou, S. P. Zhuo, Z. F. Yan, H. Gao, G. Q. Wang and S. Z. Qiao, Energy Environ. Sci., 2012, 5, 7323–7327 CAS.
- H. P. Boehm, Carbon, 1994, 32, 759–769 CrossRef CAS.
- L. Zhao, Z. Bacsik, N. Hedin, W. Wei, Y. H. Sun, M. Antonietti and M. M. Titirici, ChemSusChem, 2010, 3, 840–845 CrossRef CAS PubMed.
- M. Sevilla, P. Valle-Vigon and A. B. Fuertes, Adv. Funct. Mater., 2011, 21, 2781–2787 CrossRef CAS.
- Y. D. Xia, R. Mokaya, G. S. Walker and Y. Q. Zhu, Adv. Energy Mater., 2011, 1, 678–683 CrossRef CAS.
- S. H. Weng, Z. H. Lin, Y. Zhang, L. X. Chen and J. Z. Zhou, React. Funct. Polym., 2009, 69, 130–136 CrossRef CAS PubMed.
- L. Liu, Q. F. Deng, X. X. Hou and Z. Y. Yuan, J. Mater. Chem., 2012, 22, 15540–15548 RSC.
- L. Liu, Q. F. Deng, T. Y. Ma, X. Z. Lin, X. X. Hou, Y. P. Liu and Z. Y. Yuan, J. Mater. Chem., 2011, 21, 16001–16009 RSC.
- Y. F. Zhao, L. Zhao, K. X. Yao, Y. Yang, Q. Zhang and Y. Han, J. Mater. Chem., 2012, 22, 19726–19731 RSC.
- D. H. Lin, Y. X. Jiang, S. R. Chen, S. P. Chen and S. G. Sun, Electrochim. Acta, 2012, 67, 127–132 CrossRef CAS PubMed.
- X. P. Zhu, Y. Fu, G. S. Hu, Y. Shen, W. Dai and X. Hu, Water, Air, Soil Pollut., 2013, 224, 1387–1398 CrossRef.
- G. P. Hao, W. C. Li, D. Qian and A. H. Lu, Adv. Mater., 2010, 22, 853–857 CrossRef CAS PubMed.
- Y. Liu, J. Liu, W. Y. Yao, W. L. Cen, H. Q. Wang, X. L. Weng and Z. B. Wu, RSC Adv., 2013, 3, 18803–18810 RSC.
- H. Matsuura, H. Yoshida, M. Hieda, S. Yamanaka, T. Harada, K. Shin-ya and K. Ohno, J. Am. Chem. Soc., 2003, 125, 13910–13911 CrossRef CAS PubMed.
- S. J. Yoon, J. W. Chung, J. Gierschner, K. S. Kim, M. G. Choi, D. Kim and S. Y. Park, J. Am. Chem. Soc., 2010, 132, 13675–13683 CrossRef CAS PubMed.
- M. J. Zhong, S. Natesakhawat, J. P. Baltrus, D. Luebke, H. Nulwala, K. Matyjaszewski and T. Kowalewski, Chem. Commun., 2012, 48, 11516–11518 RSC.
- V. Chandra, S. U. Yu, S. H. Kim, Y. S. Yoon, D. Y. Kim, A. H. Kwon, M. Meyyappan and K. S. Kim, Chem. Commun., 2012, 48, 735–737 RSC.
- M. Sevilla and A. B. Fuertes, Energy Environ. Sci., 2011, 4, 1765–1771 CAS.
- J. An, S. J. Geib and N. L. Rosi, J. Am. Chem. Soc., 2010, 132, 38–39 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |