
Dissociative electron attachment to the complexation ligands hexafluoroacetylacetone, trifluoroacetylacetone and acetylacetone; a comparative experimental and theoretical study
Benedikt Ómarsson,
Sarah Engmann and
Oddur Ingólfsson*
University of Iceland, Reykjavik, Iceland. E-mail: odduring@hi.is
First published on 21st July 2014
Abstract
The β-diketones acetylacetone (AAc), trifluoroacetylacetone (TFAAc), and hexafluoroacetylacetone (HFAAc), are commonly used as ligands for metal complexes in applications where relatively high stability and vapour pressure is required. While fluorination of the native AAc generally increases both stability and vapour pressure of the respective metal complexes it also alters the electronic structure, and thereby the susceptibility to bond cleavage by low energy electrons. Here we present a detailed comparative study on dissociative electron attachment (DEA) to the isolated ligands AAc, TFAAc and HFAAc in the energy range from 0–15 eV. While single bond ruptures at fairly high energies dominate in DEA to the native AAc, extensive fragmentation, new bond formation and rearrangement is observed from the fluorinated β-diketones. These reactions have high cross sections at 0 eV, where they are often associated with stabilisation through H⋯F hydrogen bond formation and HF loss. From HFAAc considerable contributions are also observed at about 1 and 3 eV. Through comparison of the three compounds and quantum chemical calculations of the threshold energies for individual processes we are able to offer a plausible picture of the reaction dynamics behind the bulk of these channels. Finally, for the most dominating reaction channel, i.e., the loss of HF from HFAAc, we calculate the minimum energy path by using the nudged elastic band method.
1 Introduction
The β-diketone acetylacetone (AAc), and its fluorinated derivatives trifluoroacetylacetone (TFAAc) and hexafluoroacetylacetone (HFAAc) are all 2,4-pentadiones that are commonly used as complexation agents in homoleptic and heteroleptic transition metal complexes. These complexes are generally stable in air, have fairly high vapour or sublimation pressure and low decomposition temperature.1 These properties make them suitable as precursor molecules in fabrication of nano-structures and in catalyst production by thin film preparation through chemical vapour deposition (CVD).2,3 More recently such β-diketone complexes have also been considered as precursor molecules for focused electron beam induced deposition (FEBID), a promising nano-fabrication method that is based on electron-induced decomposition of the precursor molecules.4,5 In this technique the precursor molecules are decomposed at or close to the substrates surface using a highly focused, high-energy electron beam. However, when such an electron beam impinges on a surface the generation of low-energy secondary electrons (LESEs) is unavoidable.6 The reactive interaction of the precursor molecules with these low energy electrons is therefore an important parameter in the deposition process. In general the energy distribution of the LESEs peaks below 10 eV,7,8 which is below the ionisation energy of most molecules. In this energy range, dissociative electron attachment (DEA) leading to incomplete decomposition of the precursor molecules can be very efficient. Dissociative electron attachment may thus be a contributing factor in unwanted co-deposition of ligands and ligand fragments. This is of a special concern as (i) the LESEs flux is high and (ii) gas phase studies have shown that several precursor molecules used in FEBID have high DEA cross sections.9–12 It is thus important to systematically characterise the reactivity of typical and potential FEBID precursor molecules with regard to their reactivity towards low-energy electrons and use this data to better understand their role in FEBID. Such detailed studies could prove useful in establishing parameters that allow targeted synthesis of precursor molecules tailored for FEBID. In this context we have recently studied DEA to the HFAAc complexes Cu(hfac)2 and Pd(hfac)2 (ref. 12) and compared their reactivity towards low-energy electrons with that of the native ligand HFAAc. For applications where chemical stability and fairly high vapour pressure is desirable, fluorination of AAc to give TFAAc or HFAAc may favourably alter the physical and chemical properties of their respective metal complexes. Fluorination may, however, strongly influence the electronic structure of these compounds and will generally increase their susceptibility towards reactions with low energy electrons. Furthermore, in highly fluorinated compounds, where stabilisation through intramolecular H⋯F hydrogen bonds is possible, extensive fragmentation through DEA may be strongly promoted through neutral HF formation.13 Hence, the 5.8 eV bond dissociation energy (BDE) that is released in the formation of HF makes otherwise inaccessible channels thermochemically accessible. Motivated by the potential use of β-diketone complexes in FEBID and more general by the influence of fluorination on the reactivity of these compounds in their interaction with low energy electrons, we have conducted a comprehensive DEA study on HFAAc, TFAAc and AAc. We report extensive fragmentation observed in DEA to the three β-diketones and discuss these with respect to the resonances involved. For all the major fragmentation channels our discussion is supported with computed thermochemical thresholds and, in selected cases, transition state calculations along the respective reaction paths. In Section 3.1, we begin by discussing in general the nature of the resonances observed in electron attachment to the three compounds. The formation of [M − H]− and the molecular anion from TFAAc is discussed in context to the effect of fluorination on the respective resonances. Subsequently, we discuss the DEA spectra of all three compounds in two separate sections. In Section 3.2 we discuss dissociation channels leading to complex fragmentation through multiple bond ruptures from HFAAc and TFAAc. From AAc however, we only observe a single complex dissociation channel, i.e., all fragments except one, are formed through single bond rupture. We therefore limit the discussion of DEA to AAc to Section 3.3, where we discuss the single bond ruptures observed in DEA to the three β-diketones. For each major dissociation channel we discuss the reaction path suggested to dominate the reaction dynamics.2 Methods
2.1 Experimental
The experimental setup has been described in detail elsewhere14 and we therefore limit our discussion here to a short description. The experimental setup is a high-vacuum apparatus where an electron beam, formed in a trochoidal electron monochromator crosses an effusive molecular beam. The liquid samples are evaporated into the vacuum chamber through an inlet system maintained at 60 °C and the monochromator is held at a constant temperature of 120 °C. In the experiments, we assume that the temperature of the sample gas is approximately the same as the temperature of the inlet system. The base pressure for this setup is on the order of 10−8 mbar and the working pressure was maintained at approximately 5 × 10−7 mbar. The ions formed in the crossing region of the two beams are extracted by a weak (∼1 V cm−1) electric field towards a HIDEN EPIC 1000 mass spectrometer (Hiden analytical, Warrington UK). The electron energy scale was calibrated with respect to the formation of SF6− from SF6 at ∼0 eV and the electron energy resolution (120–140 meV) is estimated from the full width at half maximum (FWHM) of the SF6− signal. In the current study, the appearance energy (AE) of individual fragments is estimated through linear extrapolation of the rising side of the ion yield curve, towards the baseline. The accuracy of the reported AEs is thus affected by the electron energy resolution and we estimate error to be ∼0.2 eV. All the compounds were purchased from Sigma-Aldrich (St. Louis, MO) with a stated purity of 99% and used as delivered.2.2 Calculations
The methods used in quantum chemical calculation of thermochemical thresholds have been described elsewhere15 and we will therefore only give a brief description here. All geometry optimisations and single point energy calculations were performed using Orca 2.9 computational chemistry software.16 Harmonic vibrational frequency calculations were performed with NWChem 6.1.1.17 Thermochemical threshold values are computed on the B2PLYP18/ma-TZVP19 level of theory. The zero point vibrational energy obtained from the vibrational frequency calculations is added to each species and the 0 K thermochemical threshold, Eth, is obtained by subtracting the total energy of the fragments from the total energy of the parent molecule. To account for the experimental temperature of 60 °C, the thermally corrected threshold, E′th, is obtained by further subtracting the thermal energy of the parent molecule, at the experimental temperature, from Eth. From the vibrational frequency calculations we find this value to be 0.29, 0.35 and 0.41 eV for AAc, TFAAc and HFAAc respectively. In the gas phase at room temperature, the enol form of AAc is known to be the most stable stereoisomer.20 Additionally, the substitution of the methyl (CH3) group with CF3 has been shown to shift the keto–enol equilibrium in favour of the enol tautomer.21 This is well reflected in our calculations as we find the energy difference, at room temperature, between the two tautomers of the three β-diketones to be 0.27, 0.31 and 0.19 eV for HFAAc, TFAAc and AAc, respectively. Fig. 1 shows two possible enol conformers of TFAAc, i.e., having the CF3 group either on the keto (conformer A), or enol (conformer B) side. From our calculations we find that conformer A is 0.10 eV lower in energy than conformer B. From Boltzmann statistics this means that the enol tautomers of TFAAc mainly consist of conformer A (97% at 60 °C). While many different stereoisomers exist for the β-diketones,22,23 these all constitute a negligible portion of the gas phase molecules at the current temperature. Throughout the paper, when considering thermochemical thresholds we always refer to the enol form of the β-diketones and we always refer to the thermally corrected threshold, E′th. | ||
| Fig. 1 The two enol conformers of TFAAc. Through thermochemical calculations we find conformer A to be 101 meV lower in energy than conformer B at room temperature, and from Boltzmann statistics, 97% of the enol conformers of TFAAc are expected to exist as conformer A at 60 °C. | ||
To elucidate the dominating reaction channel, the minimum energy path for the formation and loss of HF was calculated using the nudged elastic band (NEB) method.24,25 These calculations were performed using ChemShell26 with the Orca program interfaced as QM code and the DL-FIND27 program as a geometry optimiser. The reaction path was split into three parts, i.e., the rotation of the –COCF3 group, and a two step process resulting in the formation of an anionic adduct, stabilised by hydrogen bonding between HF and the [M − HF]− moiety. Each part of the reaction path was optimised using the NEB method, with 11 images distributed along the reaction path.
3 Results and discussion
3.1 Low energy resonances in DEA to HFAAc, TFAAc and AAc; formation of M− and [M − H]−
Fig. 2 shows the ion yield curves for the loss of hydrogen ([M − H]−), from HFAAc, TFAAc and AAc, in the incident electron energy range from 0–15 eV. Fig. 3 shows the M− ion yield from TFAAc in the same energy range. The ion yield curves for the [M − H]− and the M− formation shown in Fig. 2 and 3, respectively, offer a direct comparison between the low energy contributions observed in free electron attachment to the β-diketones. In this section we thus discuss these together, while in the following sections, the more extensive fragmentation channels will be discussed for each compound separately. A part of the DEA channels observed from HFAAc have already been described previously in a study by Engmann et al.,12 however, for completeness and clarity of the text, these will also be discussed here in context to the other β-diketones. The major contributions to the ion yield observed upon DEA to HFAAc are formed through resonances close to 0, 1 and 3 eV. At these low electron energies, core-excited resonances are rarely observed and we thus attribute the contributions close to 0 and 1 eV in HFAAc, to single electron occupation of the two lowest lying molecular orbitals. The LUMO and LUMO+1 virtual orbitals resulting from the B3LYP/ma-TZVP calculations for all three β-diketones are predominately of π* (A′′) and σ* (A′) character, respectively. The same calculations, however, with the non-diffuse TZVP28 basis set, gives the LUMO and LUMO+1 order of π* and σ*, respectively, for AAc and TFAAc but π* and π*, respectively, for HFAAc. Using the even more restricted 6-31G(d) basis set results in the order π* and π* for the LUMO and LUMO+1 for all three compounds. It is thus clear that the nature of the LUMO+1 resulting from our B3LYP calculations is highly dependent on the extent of the basis set used. While it's fairly clear that the LUMO in all three molecules is of π* character we are not confident in assigning the LUMO+1. Not withstanding the symmetry of the resonances involved, one would generally expect the resonances to be shifted towards lower energy with increasing fluorination. This is the case for the second contribution in HFAAc with respect to TFAAc while the second contribution in the ion yield of AAc does not follow this trend. We cannot offer a conclusive explanation for this behaviour, but possibly this low intensity contribution is due to a different resonance than that leading to the second contribution in TFAAc and HFAAc. | ||
| Fig. 2 Ion yield curves for the formation of [M − H]− from the three β-diketones: (a) hexafluoroacetylacetone (HFAAc), (b) trifluoroacetylacetone (TFAAc) and (c) acetylacetone (AAc). | ||
 | ||
| Fig. 3 Ion yield curve for the formation of M− from trifluoroacetylacetone (TFAAc). | ||
The resonance close to 3 eV we previously assigned as a core-excited resonance associated with an optically forbidden n–π* transition.12 This transition was predicted by Nakanishi et al.29 who conducted a detailed experimental study of the electronic spectrum of the three β-diketones, supported by modified CNDO calculations. The stabilisation of the negative ion resonances (NIRs), offered by increased fluorination is clearly manifested in the relative increase of the ion yield when comparing HFAAc with TFAAc and AAc. While the [M − H]− ion yield from TFAAc and HFAAc close to 0 eV is considerable, the corresponding fragment from AAc is only observed with very low yield and slightly shifted towards higher energy. The loss of hydrogen requires the rupture of a C–H or O–H bond, both being σ bonds. In a fixed-nuclei approximation, the rupture of a σ bond can proceed directly through a σ* resonance but dissociation through a π* resonance is symmetry forbidden. In the case of π* resonances, however, molecular vibrations can lead to mixing of the occupied π* orbital with a higher lying, unoccupied σ* orbital, leading to symmetry lowering of the TNI, and thus favouring the rupture of the σ bond. This mechanism is not uncommon in DEA and has been observed in as different systems as chlorobenzene30 and acetylene.31 In the former, out-of-plane bending of the C–Cl bond leads to the required π*/σ* mixing, promoting the rupture of the C–Cl bond. In acetylene, however, the same effect is achieved through bending of the molecule. This mechanism, i.e., vibronic coupling, was also suggested to play a role in the O–H rupture of formic acid, but a recent study by Janečková et al.32 showed that in this case, the rupture of the O–H bond proceeds directly through a short-lived σ* resonance (see also ref. 33). From the orbital assignments, we suggest that the formation of [M − H]− from the three β-diketones through the 0 eV (π*) resonance is associated with coupling between the π* and the relevant σ* states of the TNI.
In the case of a reaction (such as the [M − H]− formation) that involves only one bond rupture, the thermochemical threshold is given by the BDE less the electron affinity (EA) of the charge-carrying fragment, X:
| Eth = BDE(M − X) − EA(X). | (1) |
Table 1 gives the thermochemical threshold (Eth) and the thermally corrected threshold (E′th) for the loss of hydrogen from all possible hydrogen sites of the enol form of the three β-diketones considered here (TFAAc; conformer A) along with the calculated adiabatic EAs. The EAs of the three β-diketones range from −0.21 eV (AAc) to 1.62 eV (HFAAc). For the closed shell dehydrogenated anion, we expect the HOMO to be considerably more stable than the singly occupied HOMO of the molecular anion and thus we expect the radical precursors to [M − H]− to exhibit high EAs. In fact, from our calculations we find the EA of [M − H] from HFAAc, where the hydrogen is lost from the oxygen, to be 4.13 eV. In DEA to HFAAc, if we assume the hydrogen loss to result from rupture of the O–H bond, we calculate a thermochemical threshold of −0.34 eV for the formation of [M − H]−. The reaction is thus readily accessible at 0 eV incident electron energy. If we, however, consider the H loss from the carbon atom, we find a reaction threshold of 1.27 eV. This can be explained by the facts that the C–H bond is slightly stronger than the O–H bond34 and that hydrogen loss from the oxygen ensures delocalisation of the electron in the conjugated π-system between the two oxygen atoms. The EA of the [M − H] radical is therefore expected to be considerably lower when the hydrogen is removed from the carbon atom. From Table 1, it is clear that in the case of HFAAc, the rupture of the O–H bond is energetically accessible through the full width of the 0 eV resonance while the rupture of the C–H bond is only energetically accessible above 1.27 eV. For TFAAc and AAc, the loss of hydrogen is possible from 3 molecular sites; the enol (O–H), the central carbon (C–H) and the terminal CH3 group(s) (H2C–H). For TFAAc and AAc, the formation of [M − H]− through loss of hydrogen from the oxygen is endothermic by 0.47 and 0.95 eV, respectively, while hydrogen loss from the terminal CH3 group, is endothermic by 0.74 and 1.49 eV, respectively. Finally the threshold for [M − H]− formation from TFAAc and AAc through hydrogen loss from the central carbon has a threshold of 2.09 and 3.05 eV, respectively. From these thermochemical threshold values it is clear that hydrogen loss from the oxygen of the β-diketones is the energetically most favourable process. However, despite the fact that our thermochemical calculations predict the [M − H]− formation from TFAAc and AAc to be slightly endothermic, these are observed at, or close to 0 eV. Considering the width of the electron beam and the accuracy of the calculations, the 0 eV contribution from TFAAc (E′th = 0.47 eV) might result from ground state molecules. For AAc, however, the molecules contributing to the 0 eV signal must originate from the high energy tail of the Boltzmann distribution of internal energies (E′th = 0.95 eV), i.e., hot-band transitions. We note, that though we include the temperature of the target gas in our thermally corrected thresholds, this correction does not account for the actual internal energy distribution. Another explanation can be found in considering the presence of less stable conformers than the enol considered here. If we consider the hydrogen loss from the keto tautomer of the β-diketones we derive a thermochemical threshold of −0.61, 0.17 and 0.77 eV for HFAAc, TFAAc and AAc, respectively. As mentioned above, however, we found the energy difference between the keto and enol conformers to be 0.27, 0.31 and 0.19 eV for HFAAc, TFAAc and AAc, respectively. From Boltzmann statistics this means that only ∼20, 7 and 700 ppm of HFAAc, TFAAc and AAc exist in the keto-conformer, respectively. While this may not reflect exactly, the keto–enol ratio in the vapor phase above the liquid, it is clear that if the hydrogen loss proceeds from the keto-conformer in any of the β-diketones, the DEA cross section of this conformer is quite high.
| Compound | EA | H-loss | Eth | E′th |
|---|---|---|---|---|
| HFAAc | 1.62 | O–H | 0.07 | −0.34 |
| C–H | 1.69 | 1.27 | ||
| TFAAc | 0.72 | O–H | 0.82 | 0.47 |
| H2C–H (terminal) | 1.09 | 0.74 | ||
| C–H | 2.44 | 2.09 | ||
| Ac | −0.21 | O–H | 1.25 | 0.95 |
| H2C–H (terminal) | 1.79 | 1.49 | ||
| C–H | 3.34 | 3.05 |
Interestingly, the formation of the parent anion, M−, through the 0 eV resonance is only observed from TFAAc (Fig. 3). In this case we attribute the extended lifetime, enabling observation of the molecular anion, to stabilisation through intramolecular vibrational energy redistribution (IVR). This is also manifested in the FWHM of the respective ion yield curves. The FWHM of the [M − H]− ion yield curve for TFAAc (Fig. 2b) is close to 180 meV and that of [M − HF]− is about 210 meV (discussed below). The FWHM of the parent anion contribution on the other hand, is close to 120 meV (Fig. 3). Hence, as is common for molecular anions formed in electron attachment, the parent anion can only be stabilised by IVR at very low energies and the FWHM observed, reflects the energy resolution of the electron beam. The fact that a metastable molecular anion is observed from TFAAc and not HFAAc or AAc can be rationalised by the stabilising effect offered through fluorination, increasing the EA. In fact, we calculate EAs of −0.21, 0.72 and 1.62 eV, for AAc, TFAAc and HFAAc, respectively. From the negative EA value, it is clear that AAc does not support the formation of a metastable parent anion and we thus expect autodetachment to be the dominating process. TFAAc, however, has a moderately low EA and the parent molecular anion is stabilised through IVR. For TFAAc we observe a branching ratio of IVR![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DEA of about 1:10 (maximum count rates of M− vs. total ion yield at 0 eV), i.e., DEA is more favourable than IVR for TFAAc although both are operative at 0 eV. Finally, for HFAAc, the high excess energy (EA = 1.62 eV) of the parent molecular anion formed through electron attachment at 0 eV results in unfavourable conditions for IVR, and the dominating relaxation channel (besides AD) is DEA. The parent molecular anion for HFAAc is therefore not observed within the timeframe of the current experiment. This perception is supported by comparison of the EAs of these compounds with the dissociation thresholds for the low-lying channels listed in Tables 1 and 2, respectively.
:DEA of about 1:10 (maximum count rates of M− vs. total ion yield at 0 eV), i.e., DEA is more favourable than IVR for TFAAc although both are operative at 0 eV. Finally, for HFAAc, the high excess energy (EA = 1.62 eV) of the parent molecular anion formed through electron attachment at 0 eV results in unfavourable conditions for IVR, and the dominating relaxation channel (besides AD) is DEA. The parent molecular anion for HFAAc is therefore not observed within the timeframe of the current experiment. This perception is supported by comparison of the EAs of these compounds with the dissociation thresholds for the low-lying channels listed in Tables 1 and 2, respectively.
| Fragment | m/z | Negative | Neutral | Eth | E′th | AE |
|---|---|---|---|---|---|---|
| HFAAc | ||||||
| [M − HF]− | 188 | cyclo-C5HF5O− | HF | −1.01 | −1.42 | 0 |
| C5F5O− | 171 | F3CC(O)CCCF2− | HF + OH | 3.04 | 2.63 | 4.9 |
| [M − 2HF]− | 168 | F3CCCC(O)C(O)F− | 2HF | −0.38 | −0.79 | 0 |
| cyclo-C5F4O− | 2HF | 1.41 | 1.00 | |||
| C4F5− | 143 | F3CCC(O)CF2− | CO2 + HF + H | 2.58 | 2.17 | 5.5 |
| HF + OH + CO | 3.84 | 3.43 | 0 | |||
| [M − HF − CO]− | 140 | F3CCCC(O)F− | 2HF + CO | 0.38 | −0.03 | |
| C4F3O2− | 137 | F3CC(O)CCO− | CF3 + H2 | 1.33 | 1.04 | 2.8 |
| CHF3 + H | 1.30 | 1.00 | ||||
| CF3COO− | 113 | CF3COO− | CF3CCH2 | 0.23 | −0.18 | 0 |
| C3F3H− | 94 | CF3CCH− | CF3H + CO2 | 0.87 | 0.46 | 0 |
| CF3COOH | 1.72 | 1.31 | ||||
| CF3− | 69 | CF3− | F3CCOCH2CO | 1.96 | 1.55 | 2.8 |
| HF2− | 39 | HF2− | cyclo-C5HF4O | −0.26 | −0.57 | 0 |
| F− | 19 | F− | F3CC(O)CHC(OH)CF2 | 1.35 | 0.94 | 2.9 |
| OH− | 17 | OH− | F3CC(O)CHCFCF2 | 3.49 | 3.08 | 5.4 |
| O− | 16 | O− | F3CC(O)CHCHCF3 | 3.20 | 2.91 | 5.3 |
| TFAAc | ||||||
| [M − HF]− | 134 | cyclo-C5HF5O− | HF | −0.10 | −0.45 | 0 |
| [M− 2HF]− | 114 | F3CCCC(O)C(O)F− | 2HF | 0.33 | −0.02 | 0 |
| CF3COO− | 113 | CF3COO− | CH3CCH2 | 0.45 | 0.10 | 0 |
| [M − CH3CO]− | 112 | CF3C(O)CH2− | CH3CO | 0.77 | 0.42 | 0 |
| CF3CCH− | 94 | CF3CCH− | CH4 + CO2 | 1.08 | 0.73 | 0 |
| CH3COOH | 1.65 | 1.31 | ||||
| C4H3O2− | 83 | C4H3O2− | CF3 + H2 | 2.11 | 1.76 | 3.1 |
| CH3COO− | 59 | CH3COO− | CF3CCH2 | 1.22 | 0.88 | 0 |
| CF2CCH + HF | 2.05 | 1.71 | ||||
| CF2− | 50 | CF2− | CH3COCH2COCF | 1.95 | 1.60 | 0.8 |
| CH3C(O)CHCO + HF | 2.89 | 2.54 | ||||
| CH3CCH2− | 41 | CH3CCH2− | CF3COO | 4.35 | 4.00 | 3.5 |
| CF3H + H | 2.07 | 1.72 | ||||
| HF2− | 39 | HF2− | cyclo-C5HF4O | −0.23 | −0.57 | 0 |
| F− | 19 | F− | F2CC(O)CHC(OH)CH3 | 1.46 | 1.12 | 3.2 |
| OH− | 17 | OH− | F3CC(O)CHCHCH2 | 2.39 | 2.04 | 6.2 |
| O− | 16 | O− | 3.26 | 2.92 | 4.3 | |
| AAc | ||||||
| [M − CH3CO]− | 57 | CH3COCH2− | CH3CO | 1.67 | 1.38 | 1.2 |
| CH3CO− | 43 | CH3CO− | CH3COCH2 | 3.06 | 2.76 | 7.4 |
| [M − CH3COO]− | 41 | CH3CCH2− | CH3COO | 3.87 | 3.58 | 7.1 |
| OH− | 17 | OH− | H3CCCHC(O)CH3 | 3.49 | 3.19 | 6.8 |
| O− | 16 | O− | H3CC(OH)CHCCH3 | 3.48 | 3.19 | 4.0 |
| CH3− | 15 | CH3− | H3CC(OH)CHC(O) | 3.71 | 3.42 | 7.6 |
3.2 Multiple bond ruptures and rearrangement reactions
As mentioned above, the stabilising effect of the fluorination on the TNIs observed in DEA to the title compounds is already apparent in the ion yield curves for the [M − H]− formation. This effect, however, becomes even more prominent when we consider the more complex fragmentation channels. While DEA to HFAAc is dominated by reactions with multiple bond ruptures and new bond formations, and most of these proceed already at 0 eV incident electron energy, single bond ruptures through higher-lying core-excited resonances play an increasing role in TFAAc. Finally, in AAc, single bond rupture through higher lying core-excited resonances govern the reaction dynamics and only one DEA channel is associated with multiple bond ruptures and new bond formations. To elucidate the dissociation dynamics of the complex DEA reactions, especially these that proceed close to 0 eV, we have calculated the reaction path for the most pronounced channel. We also calculated the relevant threshold energies for all DEA products observed from HFAAc. For TFAAc we calculated the threshold energies for reactions that we have identified as relevant for this discussion and for AAc we calculated the threshold energies for all DEA channels observed. Table 2 shows the 0 K thermochemical threshold, Eth, and the thermally corrected threshold E′th, calculated at the B2PLYP/ma-TZVP level of theory for the channels mentioned above. Additionally, Table 2 shows the estimated experimental AE for each of the considered fragments. | ||
| Fig. 4 Ion yield curves observed from electron attachment to HFAAc in the energy range from 0–15 eV. All the above fragments result from complex dissociation channels where bond formation and rearrangement is necessary. | ||
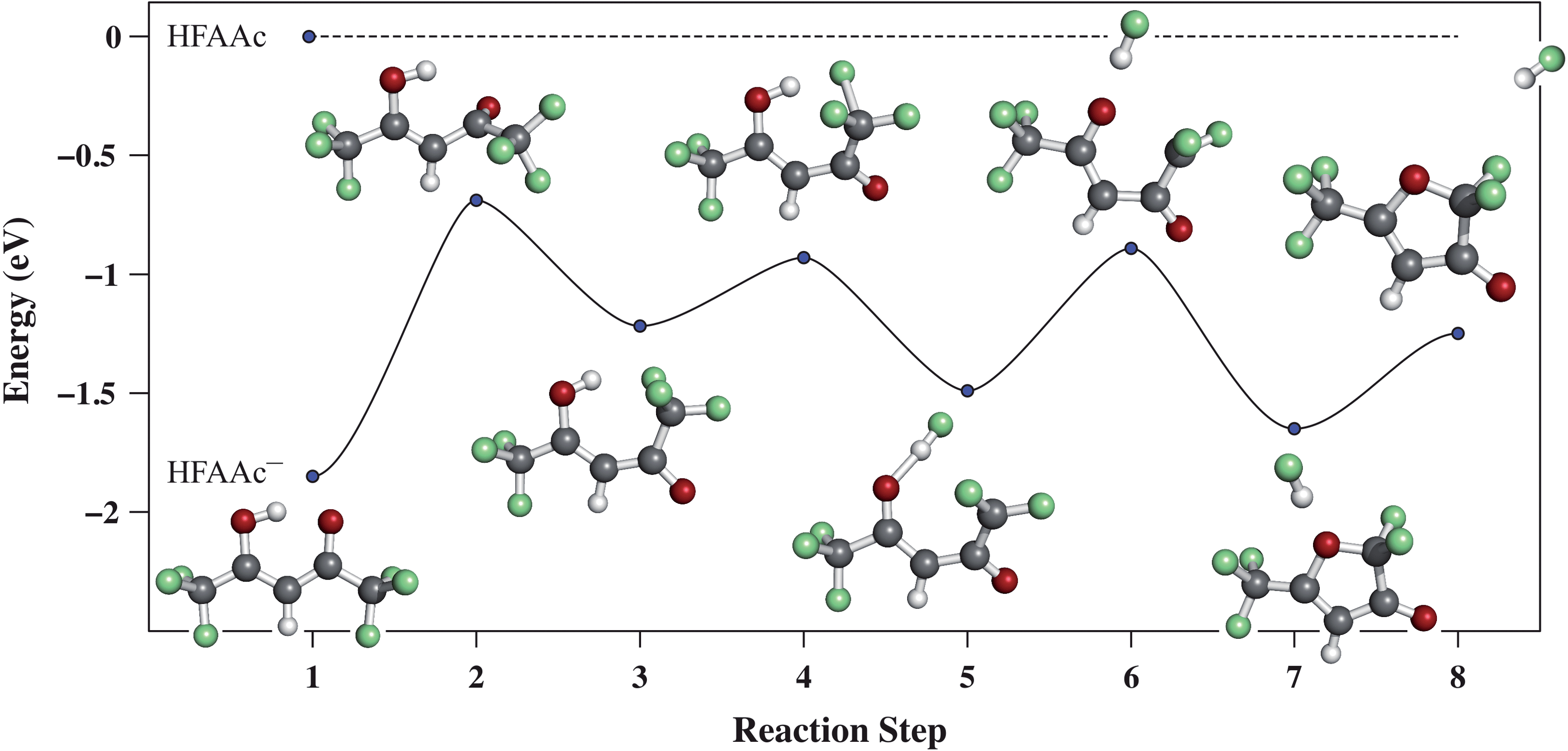 | ||
| Fig. 5 The minimum energy path for the rotation of the –COCF3 group of HFAAc− to form a –F⋯H⋯O– stabilised intermediate, followed by HF loss and concomitant ring closure. The reaction path is calculated using the nudged elastic band (NEB) method. The transition state for the rotation, which is the rate limiting step, lies energetically below the neutral HFAAc, making this rotation readily available at 0 eV incident electron energy. In analogy to the mechanism proposed by Basset and Whittle,35 this rotation is proposed as a prerequisite to the loss of HF upon DEA. | ||
Here, similar to our observations for PFP and PFA13,38 the energy released through the formation of HF (∼5.8 eV) promotes dissociation channels, otherwise energetically inaccessible. In fact, a number of the dissociation channels observed upon electron attachment to HFAAc (see Table 2 and Fig. 4) are only energetically accessible through HF bond formation. The formation of [M − 2HF]− (Fig. 4b) formed through the 0, 1 and the 3 eV resonance, is an example of such a channel. In this case, a total of four chemical bonds are broken, a process that requires considerable energy. According to our thermochemical calculations, however, the formation of two HF may lead to a threshold that is sufficiently low for this process to proceed at 0 eV, where this fragment is observed. Originally, we expected this reaction path to involve the loss of one fluorine from each CF3 group. However, the formation of [M − 2HF]− is also observed from TFAAc (see Section 3.2.2), in which case both fluorines are bound to come from the same CF3 group. If we assume that this reaction follows a similar path as the loss of a single HF molecule, i.e., initial rotation of the –COCF3 group after electron capture, HF loss and the formation of a 5-membered ring similar to the furanone, the calculated threshold is about 1 eV. If we assume, however, that rather than closing the ring, the reaction proceeds through a path as depicted in Fig. 6, we derive a threshold of −0.79 eV. We note that the first structure of the intermediates shown in Fig. 6 represents an intermediate from the path calculated for the loss of one HF unit (step 3 in Fig 5), while the second structure is only tentative and does not necessarily represent a minimum on the actual reaction path.
 | ||
| Fig. 6 Proposed reaction path and final product for the formation of [M − 2HF]− from HFAAc. Assuming the final product shown in the figure, the thermochemical threshold is found to be −0.79 eV. | ||
With an ion yield profile, similar to that of [M − 2HF]− we also observe the fragment [M − 2HF − CO]− (Fig. 5c). This fragment is, however, formed in higher yield through the resonances close to 1 and 3 eV than through the 0 eV resonance, from which the [M − 2HF]− formation is strongest. We take this as an indication that [M − 2HF − CO]− is formed on the same or a similar reaction path as anticipated for [M − 2HF]−. Hence, for this channel to proceed, an additional C–C bond must be ruptured and the fluorine must transfer to the carbonyl group next to the allyl group in the final structure in Fig. 6. Thus we expect, as observed, that the branching ratio for this channel is favoured with increasing internal energy, i.e., through the higher-lying resonances. This is also supported by the threshold energy we derive for this process by assuming the formation of two HFs, CO and CF3–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–CFO−, i.e., −0.03 eV. Such extensive fragmentation as we observe here is not unique and has been observed before in DEA to tetrafluoro-p-benzoquinone15 where the excision of both CO groups from the quinone ring, followed by extensive rearrangement and recombination of the C2 units, leads to the formation of the linear C4− anion and two neutral F2CO molecules.
C–CFO−, i.e., −0.03 eV. Such extensive fragmentation as we observe here is not unique and has been observed before in DEA to tetrafluoro-p-benzoquinone15 where the excision of both CO groups from the quinone ring, followed by extensive rearrangement and recombination of the C2 units, leads to the formation of the linear C4− anion and two neutral F2CO molecules.
A quite interesting fragment, HF2−, is also formed in DEA to HFAAc through the 0 eV resonance (see Fig. 4d). This fragment, known as bifluoride, is a linear and symmetric anion with a 3-center, 4-electron bond between a central hydrogen and two terminal fluorines. The HF2− anion has previously been observed in DEA to difluoroethylenes39 and trifluoroacetone.40,41 In HFAAc, this reaction channel requires the rupture of two C–F bonds and a single O–H bond whereas energy is gained from the F–H–F bond formation, and from the considerable EA of HF2. This value has been estimated to be close to 4.8 eV.39 In order to account for the formation of HF2− through the 0 eV resonance we have considered a similar reaction path as for the loss of HF. In this case, however, additional fluorine is lost from the same CF3 group and the reaction leads to the formation of a furanone that has one fluorine less than that formed in the case of HF loss. The thermochemical threshold obtained for this channel is −0.57 eV, supporting this reaction path at 0 eV. Further fragments observed close to 0 eV in DEA to HFAAc, are CF3CCH− (Fig. 4e) and the trifluoroacetate anion, CF3COO− (Fig. 4f). From a stoichiometric point of view, these fragments are complementary with regard to the retention of the hydrogen atom, and both are also formed through the resonances close to 1 and 3 eV. However, for these fragments the branching ratio is about 10/1 in favour of CF3CCH−, and while the 0 eV contribution dominates in the case of CF3COO−, the contributions through the 0 and 1 eV resonance for CF3CCH− are of similar yield. Furthermore, the formation of CF3COO− is not obvious from the enol conformer of HFAAc, as it requires transferring the oxygen of the hydroxyl group to the carboxyl carbon and the hydrogen of the hydroxyl group to the central carbon. Nonetheless, the thermochemical threshold for this channel is −0.18 eV if we assume the complementary neutral fragment to be CF3CCH2. The formation of the trifluoroacetate, CF3COO− is therefore thermochemically accessible at 0 eV incident electron energy given that the reaction path proceeds through transition states lower in energy than the neutral HFAAc molecule. In the case of CF3CCH−, if we assume that CF3COOH is the complementary fragment (with regard to the hydrogen retention) we calculate a threshold of 1.31 eV, and thus not accessible through the 0 eV resonance. If instead, this DEA reaction leads to the neutral fragments CO2 and trifluoromethane, CF3H, the thermochemical threshold is lowered to 0.46 eV. This is still higher than the observed AE, but can be rationalised by considering the energy spread of the electron beam, the fact that our thermally corrected thresholds do not account for the internal energy distribution, and the lack of possible reaction paths with lower threshold. We thus suggest that the formation of this fragment at 0 eV is promoted through the formation of neutral CO2 and trifluoromethane, CF3H. We note that the formation of carbon dioxide has been observed in earlier surface-DEA experiments to small organic acids,42 we are, however, not aware of any such studies on β-diketones. We also note that similar to the case of [M − H]− from TFAAc and AAc, if this reaction proceeds from the keto-form of HFAAc the threshold is lowered by about 0.3 eV.
In addition to the fragments formed close to 0 eV, and through the resonances close to 1 and 3 eV, we observe C4F3O2− (Fig. 4g), formed through a core-excited resonance close to 3.5 eV and through overlapping core-excited resonances in the range 5–9 eV. Furthermore, C5F5O− (Fig. 4h) and C4F5− (Fig. 4i) are formed through core-excited resonances centred around 6 and 7 eV, respectively. These are most likely the same resonances that yield C4F3O2− between 5 and 9 eV. Despite the comparatively high appearance energy of these fragments they cannot be formed through simple bond ruptures, but all require rearrangement and new bond formation. In turn, however, due to their comparatively high appearance energies our thermochemical threshold calculations cannot offer any conclusive insight into the nature of the fragmentation processes. We thus only list, in Table 2, the lowest thermochemical thresholds we have calculated for these and refrain from further discussion here.
Fig. 7 shows the ion yield curves obtained from DEA to TFAAc where the dissociation channels are governed by multiple events, involving bond ruptures, bond formations and rearrangement. Same as for HFAAc, the dominating contribution is [M − HF]− (Fig. 7a) observed close to 0 eV incident electron energy. For TFAAc we calculate a threshold of −0.45 eV for this reaction. The structure of the favoured TFAAc conformer (see conformer A in Fig. 1) supports rotation of the –COCF3 group after electron capture, i.e., rotation is not hindered by a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond. We thus expect the reaction path to be similar to that of HFAAc, i.e., rotation to a hydrogen bonded intermediate promoting HF loss and the formation of [M − HF]− in the furanone form. In fact this reaction was also observed for neutral TFAAc in a recent photolysis study by Muyskens et al.37 From TFAAc, we also observe [M − 2HF]− (Fig. 7b) and HF2− (Fig. 7c) close to 0 eV and we calculate the threshold for these reactions to be −0.02 and −0.57 eV respectively. Unlike HFAAc, however, the formation of these fragments from TFAAc can only result through the loss of two fluorines from the same CF3 group. We thus also expect that both fluorines involved in the formation of HF2− and [M − 2HF]− from HFAAc are from the same CF3 group, as discussed above. Hence, at around 0 eV, we expect these reactions to proceed through similar paths in TFAAc and HFAAc (see Fig. 5 and 6). In addition to the 0 eV contribution, the formation of HF2− is observed through minor contributions around 4 and from 7–12 eV. The HF2− formation through these high-energy core-excited resonances may also be associated with the formation of a furanone, but such rearrangement is not necessary at these energies. In addition to the fragments discussed here above, trifluoroacetate (CF3COO−), acetate (CF3COO−) and CF3CCH−, are also observed from TFAAc, at or close to 0 eV (see Fig. 7d–f). From these, CF3COO− is the only fragment with appreciable yield, formed through the higher lying core-excited resonances, at about 4 and 8 eV. The reaction path, leading to the formation of CF3COO− is expected to be similar to that for HFAAc and the thermochemical threshold is found to be 0.10 eV if we assume the neutral fragment CF3CCH2. The anionic counterpart, CH3CCH2− (Fig. 7g) (complementary to CF3COO−), is only formed through the higher lying core-excited resonances with low yield from slightly below 4 to about 6 eV and from about 6–12 eV. Assuming the same reaction path as for the formation of CF3COO−, with the charge retention at the CF3CCH2 moiety, we find the thermochemical threshold to be 4.0 eV, which is in fairly good agreement with the ion yield curves observed. If the formation of CH3COO− from the most stable conformer of TFAAc proceeds in a similar way to the CF3COO− formation, now with a neutral CF3CCH− fragment, we derive a threshold of 0.88 eV. We also considered the formation of HF and a CF2CCH radical, but this channel we find to be endothermic by 1.71 eV. Finally, for CF3CCH−, similar to its formation from HFAAc, we derive a threshold of 1.31 eV if we assume the neutral fragment to be CH3COOH. However, considering the formation of CO2 and methane CH4 results in a threshold of 0.73 eV. The threshold values we calculate for CF3CCH− and for CF3COO− are markedly higher than the observed AE. This might be explained by considering the keto tautomer or hot-band transitions, as mentioned above.
C double bond. We thus expect the reaction path to be similar to that of HFAAc, i.e., rotation to a hydrogen bonded intermediate promoting HF loss and the formation of [M − HF]− in the furanone form. In fact this reaction was also observed for neutral TFAAc in a recent photolysis study by Muyskens et al.37 From TFAAc, we also observe [M − 2HF]− (Fig. 7b) and HF2− (Fig. 7c) close to 0 eV and we calculate the threshold for these reactions to be −0.02 and −0.57 eV respectively. Unlike HFAAc, however, the formation of these fragments from TFAAc can only result through the loss of two fluorines from the same CF3 group. We thus also expect that both fluorines involved in the formation of HF2− and [M − 2HF]− from HFAAc are from the same CF3 group, as discussed above. Hence, at around 0 eV, we expect these reactions to proceed through similar paths in TFAAc and HFAAc (see Fig. 5 and 6). In addition to the 0 eV contribution, the formation of HF2− is observed through minor contributions around 4 and from 7–12 eV. The HF2− formation through these high-energy core-excited resonances may also be associated with the formation of a furanone, but such rearrangement is not necessary at these energies. In addition to the fragments discussed here above, trifluoroacetate (CF3COO−), acetate (CF3COO−) and CF3CCH−, are also observed from TFAAc, at or close to 0 eV (see Fig. 7d–f). From these, CF3COO− is the only fragment with appreciable yield, formed through the higher lying core-excited resonances, at about 4 and 8 eV. The reaction path, leading to the formation of CF3COO− is expected to be similar to that for HFAAc and the thermochemical threshold is found to be 0.10 eV if we assume the neutral fragment CF3CCH2. The anionic counterpart, CH3CCH2− (Fig. 7g) (complementary to CF3COO−), is only formed through the higher lying core-excited resonances with low yield from slightly below 4 to about 6 eV and from about 6–12 eV. Assuming the same reaction path as for the formation of CF3COO−, with the charge retention at the CF3CCH2 moiety, we find the thermochemical threshold to be 4.0 eV, which is in fairly good agreement with the ion yield curves observed. If the formation of CH3COO− from the most stable conformer of TFAAc proceeds in a similar way to the CF3COO− formation, now with a neutral CF3CCH− fragment, we derive a threshold of 0.88 eV. We also considered the formation of HF and a CF2CCH radical, but this channel we find to be endothermic by 1.71 eV. Finally, for CF3CCH−, similar to its formation from HFAAc, we derive a threshold of 1.31 eV if we assume the neutral fragment to be CH3COOH. However, considering the formation of CO2 and methane CH4 results in a threshold of 0.73 eV. The threshold values we calculate for CF3CCH− and for CF3COO− are markedly higher than the observed AE. This might be explained by considering the keto tautomer or hot-band transitions, as mentioned above.
 | ||
| Fig. 7 Ion yield curves observed from electron attachment to TFAAc in the energy range from 0–15 eV. All the above fragments result from complex dissociation channels where bond formation and rearrangement is necessary. | ||
Finally the fragments CF2− (Fig. 7h) and C4H3O2− (Fig. 7i) are observed from TFAAc. Both are formed through the core-excited resonances close to 4 eV and between 6 and 10 eV, and CF2− is also formed between 1 and 2 eV. The formation of CF2− requires the rupture of the C–CF3 bond and the additional loss of a fluorine. The lowest threshold we find for this fragment is 1.60 eV and includes formation of acetoacetyl fluoride (CH3COCH2C(O)F). By assuming the formation of CF2− involves the formation of HF and acetylketene (CH3C(O)CHCO) we, however, find the threshold to be 2.45 eV. The fragment C4H3O2−, on the other hand, has a fairly high AE and can be associated with the formation of trifluoromethane (CF3H), but also the formation of CF3 and H2 results in a threshold well below the observed AE (see Table 2).
3.3 Single bond ruptures
From the fragments formed through the rupture of a single bond upon DEA to HFAAc, TFAAc and AAc, only the fragments OH− and O− are observed from all three compounds. For this reason and the fact that the OH− yield reflects the degree of fluorination of the three β-diketones very clearly, we will discuss these before discussing the remaining fragment for each β-diketone separately. Fig. 8 shows the OH− ion yield curves from HFAAc, TFAAc and AAc in panel (a), (b) and (c), respectively, and that of O− in panels (d), (e) and (f), respectively. The formation of OH− is observed through a single contribution which shape and width is similar for all compounds. The onset and the maximum, on the other hand, is successively shifted to higher energy when proceeding from HFAAc through TFAAc to AAc. In the ion yield curve for HFAAc the maximum is close to 7.4 eV, in that for TFAAc close to 8.1 eV and from AAc close to 8.8 eV. Similar trend is observed for the onset of this contribution. In a previous photochemical study, Yoon et al. observed the spontaneous loss of OH from HFAAc (and AAc), upon electronic excitation through photon irradiation in the range from 312–290 nm (3.98–4.28 eV).43 The authors attributed the excitation to a π–π* transition observed as a broad structure, centred at 4.72 eV in the UV spectrum.29 In a later photodissociation study on AAc, Upadhyaya et al.44 observed the loss of OH at 193 nm (6.43 eV), which they attributed to a dissociation resulting from an excitation to a σ* repulsive state. In accordance with the photochemical study by Upadhyaya et al.44 we attribute the formation of OH− in DEA to HFAAc, to a core-excited σ*-type resonance from the enolic part of HFAAc. This contribution is successively shifted to lower energy as the degree of fluorination increases. We note, however, that we calculate the thermochemical threshold for the formation of OH− from all three β-diketons, to be slightly above 3 eV. Thus, in principle this channel should be accessible in the energy range where Yoon et al.43 observe OH loss in their photodissociation study, i.e., around 4 eV. We cannot offer any definitive reason for the lack of OH− formation in this energy range, especially for HFAAc and TFAAc where other fragments are observed in this energy range. However, a possible explanation can be in the high activation barrier we expect for OH− loss from the enolic form due to the hydrogen bond.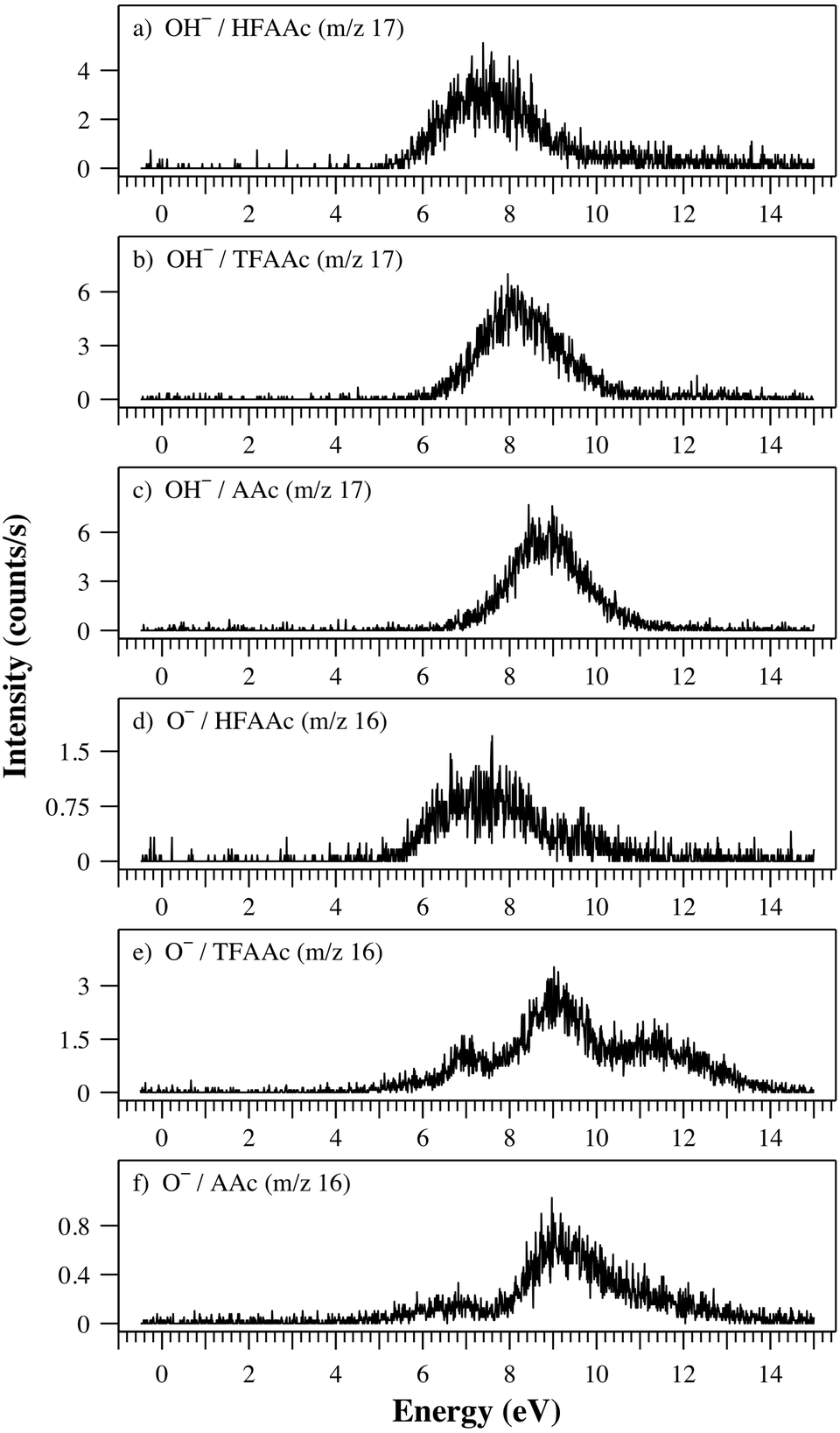 | ||
| Fig. 8 Ion yield curves for the formation of OH− and O− from HFAAc, TFAAc and AAc in the energy range from 0–15 eV. | ||
The formation of O−, from all three compounds, is observed through multiple contributions, centred around 7, 9 and 11.5 eV. The 7 eV contribution is by far the dominating one from HFAAc while the 9 eV contribution dominates from both TFAAc and AAc. Finally the 11.5 eV contributions is only significant in the O− ion yield from TFAAc. In our calculations, optimisation of the neutral [M − O] results in hydrogen transfer from the enolic oxygen to the carbon atom that contained the keto oxygen and the thermochemical threshold for this channel is found to be about 3 eV for all three β-diketones. We attribute the formation of O− from HFAAc to result from multiple core-excited π* and σ* type resonances.
 C and C–C bond ruptures at the enol and keto side of TFAAc, respectively. In TFAAc, the main contributions to the ion yields of CF−3 and F− are formed through resonances close to 8 eV and a lesser contribution is observed close to 4 eV. In the vacuum UV spectrum of TFAAc, an n–σ* transition is observed at 9.7 eV (ref. 29) but in DEA to TFAAc, we do not observe the formation of CF3− from a core-excited resonance in this energy range, and F− is only formed with low intensity close to 10.5 eV. This is in contrast to HFAAc (see Fig. 9) where a prominent contribution to the ion yields of F− and CF3− is observed close to 10.5 eV. We attribute this to a lesser stabilisation of the 10.5 eV resonance in TFAAc, due to lower degree of fluorination, thus allowing autodetachment to compete more effectively with dissociation in this energy range. We find the thermochemical threshold for this channel to be 0.42 eV. If we consider this reaction to take place from the keto tautomer, however, the threshold is lowered to 0.12 eV and we would also expect the activation barriers to be lower. The complementary fragment, CH3CO−, is also formed, but only with low intensity and through the core-excited resonances close to 4, 8 and 11 eV. We have not calculated the threshold for this fragment explicitly for TFAAc but for AAc we find a threshold of 2.76 eV and we expect the threshold to be similar for TFAAc. Finally, the fragment CF3CO− is formed through the core-excited resonance around 8 eV and the complementary fragment, [M − CF3CO]−, is observed around 4, 8 and 11 eV. The ion yield of both these fragments is, however, very low.
C and C–C bond ruptures at the enol and keto side of TFAAc, respectively. In TFAAc, the main contributions to the ion yields of CF−3 and F− are formed through resonances close to 8 eV and a lesser contribution is observed close to 4 eV. In the vacuum UV spectrum of TFAAc, an n–σ* transition is observed at 9.7 eV (ref. 29) but in DEA to TFAAc, we do not observe the formation of CF3− from a core-excited resonance in this energy range, and F− is only formed with low intensity close to 10.5 eV. This is in contrast to HFAAc (see Fig. 9) where a prominent contribution to the ion yields of F− and CF3− is observed close to 10.5 eV. We attribute this to a lesser stabilisation of the 10.5 eV resonance in TFAAc, due to lower degree of fluorination, thus allowing autodetachment to compete more effectively with dissociation in this energy range. We find the thermochemical threshold for this channel to be 0.42 eV. If we consider this reaction to take place from the keto tautomer, however, the threshold is lowered to 0.12 eV and we would also expect the activation barriers to be lower. The complementary fragment, CH3CO−, is also formed, but only with low intensity and through the core-excited resonances close to 4, 8 and 11 eV. We have not calculated the threshold for this fragment explicitly for TFAAc but for AAc we find a threshold of 2.76 eV and we expect the threshold to be similar for TFAAc. Finally, the fragment CF3CO− is formed through the core-excited resonance around 8 eV and the complementary fragment, [M − CF3CO]−, is observed around 4, 8 and 11 eV. The ion yield of both these fragments is, however, very low.

 | ||
| Fig. 11 Ion yield curves from DEA to acetylacetone (AAc) in the energy range from 0–15 eV. Apart from panel (d) which represents a complex dissociation channel, all the fragments can be formed through single bond rupture. In addition to the above fragments, [M − H]− (Fig. 1), OH− and O− (Fig. 8) are also observed. | ||
Finally, the fragment [M − CH3COO]− is formed through the core-excited resonance close to 9 eV. This is the only fragment we observe from AAc that requires multiple bond ruptures and new bond formation. Considering the anion CH3CCH2− and the neutral counterpart CH3COO we derive a threshold of 3.58 eV, which is well below the observed AE. The loss of CH3COO was not observed in DEA to TFAAc, although we did observe [M − CF3COO]−, hence the formation of the anion CH3CCH2−. Interestingly, from HFAAc where this fragment (CH3CCH2−) cannot be formed, the corresponding loss of CF3COO is not observed.
In addition, to the fragments discussed above, we have also looked for H− formation from AAc. While we cannot efficiently extract H− in our current experimental setup due to the high magnetic field, we were still able to detect H− from AAc through the high-energy resonances. Due to large background signal, however, and a large error in the electron energy due to the strong extraction field needed to extract the H− ion, we feel the data is ill suited for publication at this stage and we refrain from further discussion on the energy dependence of the H− formation.
4 Conclusions
In low energy electron interaction with the β-diketones; acetylacetone, trifluoroacetylacetone and hexafluoroacetylacetone electron attachment leads predominantly to fragmentation and within the time window of the current experiments, the molecular anion is only observed from TFAAc. The degree of fluorination is clearly reflected in the dissociation reactions of the respective compounds, (i) through stabilisation of the NIRs involved and (ii) through facilitation of fragmentation by HF formation. The second effect, in conjunction with quantum chemical calculations, allows us to explain the reaction paths behind the formation of a number of the fragments observed. While DEA to AAc is characterised by single bond ruptures that proceed with low cross sections through core-excited resonances at fairly high energy (>4 eV), DEA to TFAAc leads to about the same number of fragments through single bond ruptures as through complex rearrangement reaction. The rearrangement reactions, however, proceed mainly at low energies and generally with considerably higher cross sections. Finally, DEA to HFAAc is dominated by complex rearrangement reactions through the low energy resonances, both with respect to the number of fragments and the cross section for the individual processes. The main channel in DEA to both TFAAc and HFAAc is the formation of HF but also the loss of two HFs and the formation of HF2− are observed, though with considerably less intensity. Our threshold and nudged elastic band (NEB) calculations show that the channels leading to the loss of HF are enabled through an O⋯H⋯F intermediate leading to the formation of a furanone-like compound concomitant to the HF loss. While HF2− formation most likely proceeds through a similar path, the loss of two HFs, leads to an open chain anion. It is worth noting that the HF2− formation and the loss of two HFs proceed to similar extent from TFAAc and HFAAc, and both fluorines lost through the respective channels are thus likely to be from the same CF3 group. The main contribution to the ion yield from HFAAc and TFAAc is observed close to 0 eV, while considerable contributions from HFAAc are also observed close to 1 and 3 eV.It is clearly demonstrated here that the degree of fluorination increases the DEA cross section considerably, a phenomena that is fairly well understood and is common to most organic compounds. However, in conjunction to our previous studies, it is also clear that by introducing the stereochemical prerequisites for an energetically accessible hydrogen bonded X⋯H⋯F intermediate on the DEA reaction path, molecular fragmentation may be significantly enhanced through HF formation. We thus propose that introducing such predetermined breaking points into molecular systems, may be used to increase the molecules' sensitivity to bond breakage by low energy electrons and even control their reactivity towards these. As pointed out in the introduction, DEA can be disadvantageous in FEBID as secondary (back-scattered) electrons can contribute to deposit-impurities through incomplete dissociation of the precursor molecules. However, we point out that the main fragmentation channels we observe in the current study are promoted through the formation of HF. In a recent study by Engmann et al.,12 these reaction channels were shown to be closed when the deprotonated HFAAc is bound as a bidendate ligand in the metal-complexes Cu(hfac)2 and Pd(hfac)2.
Enhancing the DEA fragmentation efficiency of radiosensitisers used in cancer therapy may also prove beneficial. Halogenated radio sensitisers such as 5-halouracils48 have been proposed to owe their effectiveness to increased electron attachment cross section leading to low energy electron induced damaged to the DNA. Increasing the DEA cross section of radiosensitisers through the introduction of predetermined breaking points, could thus increase the effectiveness of radiosensitisers in ion beam cancer therapy.
Acknowledgements
The authors acknowledge financial support from the Icelandic Centre for Research (RANNÍS) and the University of Iceland Research Fund. We would like to thank Birkir Reynisson for his contribution to the measurements and Frímann Haukur Ómarsson for estimating the appearance energies. This work was conducted within the framework of the COST actions MP1002 on Nanoscale Insights into Ion Beam Cancer Therapy and CM1301 on Chemistry for Electron-Induced Nano-fabrication.References
- R. E. Sievers and J. E. Sadlowski, Science, 1978, 201, 217–223 CAS.
- J.-Y. Zhang and H. Esrom, Appl. Surf. Sci., 1992, 54, 465–470 CrossRef CAS.
- T. Maruyama and T. Shirai, J. Mater. Sci., 1995, 30, 5551–5553 CrossRef CAS.
- I. Utke, P. Hoffmann and J. Melngailis, J. Vac. Sci. Technol., B: Microelectron. Nanometer Struct.–Process., Meas., Phenom., 2008, 26, 1197–1276 CrossRef CAS.
- W. F. Van Dorp and C. W. Hagen, J. Appl. Phys., 2008, 104, 081301 CrossRef PubMed.
- N. Silvis-Cividjian, C. W. Hagen and P. Kruit, J. Appl. Phys., 2005, 98, 084905 CrossRef PubMed.
- T. Koshikawa and R. Shimizu, J. Phys. D: Appl. Phys., 1973, 6, 1369 CAS.
- J. Schaefer and J. Hoelzl, Thin Solid Films, 1972, 13, 81–86 CrossRef CAS.
- S. Engmann, M. Stano, S. Matejčík and O. Ingólfsson, Angew. Chem., Int. Ed., 2011, 50, 9475–9477 CrossRef CAS PubMed.
- O. May, D. Kubala and M. Allan, Phys. Chem. Chem. Phys., 2012, 14, 2979 RSC.
- S. Engmann, M. Stano, S. Matejčík and O. Ingólfsson, Phys. Chem. Chem. Phys., 2012, 14, 14611–14618 RSC.
- S. Engmann, B. Ómarsson, M. Lacko, M. Stano, S. Matejčík and O. Ingólfsson, J. Chem. Phys., 2013, 138, 234309 CrossRef PubMed.
- B. Ómarsson, E. H. Bjarnason, O. Ingólfsson, S. Haughey and T. A. Field, Chem. Phys. Lett., 2012, 539–540, 7–10 CrossRef PubMed.
- E. H. Bjarnason, B. Ómarsson, S. Engmann, F. H. Ómarsson and O. Ingólfsson, Eur. Phys. J. D, 2014, 68, 121 CrossRef.
- B. Ómarsson and O. Ingólfsson, Phys. Chem. Chem. Phys., 2013, 15, 16758–16767 RSC.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 2, 73–78 CrossRef.
- M. Valiev, E. J. Bylaska, N. Govind, K. Kowalski, T. P. Straatsma, H. J. J. Van Dam, D. Wang, J. Nieplocha, E. Apra, T. L. Windus and W. A. de Jong, Comput. Phys. Commun., 2010, 181, 1477–1489 CrossRef CAS PubMed.
- L. Goerigk and S. Grimme, J. Chem. Theory Comput., 2011, 7, 291–309 CrossRef CAS.
- J. Zheng, X. Xu and D. G. Truhlar, Theor. Chem. Acc., 2010, 128, 295–305 CrossRef.
- H. Nakanishi, Bull. Chem. Soc. Jpn., 1977, 50, 2255–2261 CrossRef CAS.
- J. L. Burdett and M. T. Rogers, J. Am. Chem. Soc., 1964, 86, 2105–2109 CrossRef CAS.
- N. Nagashima, S. Kudoh and M. Nakata, Chem. Phys. Lett., 2003, 374, 59–66 CrossRef CAS.
- S. Coussan, Y. Ferro, A. Trivella, M. Rajzmann, P. Roubin, R. Wieczorek, C. Manca, P. Piecuch, K. Kowalski, M. Włoch, S. A. Kucharski and M. Musiał, J. Phys. Chem. A, 2006, 110, 3920–3926 CrossRef CAS PubMed.
- H. Jónsson, G. Mills and K. W. Jacobsen, Classical and Quantum Dynamics in Condensed Phase Simulations, World Scientific, Singapore, 1998, p. 385 Search PubMed.
- H. Jónsson, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 944–949 CrossRef PubMed.
- Chemshell, a Computational Chemistry Shell, see: http://www.chemshell.org.
- J. Kästner, J. M. Carr, T. W. Keal, W. Thiel, A. Wander and P. Sherwood, J. Phys. Chem. A, 2009, 113, 11856–11865 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- H. Nakanishi, H. Morita and S. Nagakura, Bull. Chem. Soc. Jpn., 1978, 51, 1723–1729 CrossRef CAS.
- T. Skalický, C. Chollet, N. Pasquier and M. Allan, Phys. Chem. Chem. Phys., 2002, 4, 3583–3590 RSC.
- R. Dressler and M. Allan, J. Chem. Phys., 1987, 87, 4510 CrossRef CAS PubMed.
- R. Janečková, D. Kubala, O. May, J. Fedor and M. Allan, Phys. Rev. Lett., 2013, 111, 213201 CrossRef.
- G. Gallup, P. Burrow and I. Fabrikant, Phys. Rev. A, 2009, 79, 042701 CrossRef.
- J. R. B. Gomes and M. A. V. R. da Silva, J. Phys. Chem. A, 2006, 110, 13948–13955 CrossRef CAS PubMed.
- J. E. Bassett and E. Whittle, Int. J. Chem. Kinet., 1976, 8, 859–876 CrossRef CAS.
- G. C. Pimentel, F. M. G. Tablas, J. Hartmann and E. Whittle, Int. J. Chem. Kinet., 1976, 8, 877–882 CrossRef CAS.
- K. J. Muyskens, J. R. Alsum, T. A. Thielke, J. L. Boer, T. R. Heetderks and M. A. Muyskens, J. Phys. Chem. A, 2012, 116, 12305–12313 CrossRef CAS PubMed.
- B. Ómarsson, E. H. Bjarnason, S. A. Haughey, T. A. Field, A. Abramov, P. Klüpfel, H. Jónsson and O. Ingólfsson, Phys. Chem. Chem. Phys., 2013, 15, 4754–4766 RSC.
- M. Heni and E. Illenberger, J. Chem. Phys., 1985, 83, 6056 CrossRef CAS PubMed.
- T. Oster, PhD thesis, Freie Universität Berlin, 1987.
- E. Illenberger and M. Meinke, J. Phys. Chem. A, 2014 DOI:10.1021/jp503129z.
- I. Martin, M. Bertin, A. Domaracka, R. Azria, E. Illenberger and A. Lafosse, Int. J. Mass Spectrom., 2008, 277, 262–268 CrossRef CAS PubMed.
- M.-C. Yoon, Y. S. Choi and S. K. Kim, Chem. Phys. Lett., 1999, 300, 207–212 CrossRef CAS.
- H. P. Upadhyaya, A. Kumar and P. D. Naik, J. Chem. Phys., 2003, 118, 2590 CrossRef CAS PubMed.
- I. Bald, J. Langer, P. Tegeder and O. Ingólfsson, Int. J. Mass Spectrom., 2008, 277, 4–25 CrossRef CAS PubMed.
- O. Ingólfsson, F. Weik and E. Illenberger, Int. Rev. Phys. Chem., 1996, 15, 133–151 CrossRef.
- M. R. Nimlos, J. A. Soderquist and G. B. Ellison, J. Am. Chem. Soc., 1989, 111, 7675–7681 CrossRef CAS.
- H. Abdoul-Carime, M. A. Huels, E. Illenberger and L. Sanche, J. Am. Chem. Soc., 2001, 123, 5354–5355 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |