DOI:
10.1039/C4RA04444D
(Paper)
RSC Adv., 2014,
4, 26063-26073
Molecularly imprinted layer-coated hollow polysaccharide microcapsules toward gate-controlled release of water-soluble drugs
Received
12th May 2014
, Accepted 2nd June 2014
First published on 4th June 2014
Abstract
Molecularly imprinted polymers (MIPs) as drug delivery carriers have been explored, but it still remains a significant challenge to achieve a high drug loading amount and controlled drug release rate of MIPs. Here, we report a layer-by-layer self-assembly method for doxorubicin (DOX) imprinting of biocompatible microcapsules, which consist of carboxymethyl cellulose–(chitosan/alginate)5 and are able to encapsulate and deliver DOX. It has been demonstrated that the maximum loading capacity of the obtained hollow molecularly imprinted microcapsules (MIMs) to DOX was up to 155.1 μmol g−1. Uncommonly, the release process of DOX-loaded MIMs could be sustained for more than 168 h, resulting from the imprinted sites being blocked by DOX molecules, which entered the imprinted sites through electrostatic interaction. Moreover, the interaction could be weakened at lower pH due to the protonation of the carboxyl groups of O-carboxymethyl chitosan, leading to the DOX departing from the imprinted sites (namely DOX release). Meanwhile, in vitro experiments demonstrated that the hollow DOX-imprinted microcapsules had good biocompatibility, the encapsulated DOX could effectively induce the apoptosis of U373 malignant glioma cells and had better efficacy than that of free DOX in terms of tumor inhibition within 144 h. These findings indicate that the construction of the novel smart microcapsules of natural polymers plays a crucial role in the performance of controlled drug release. This facile strategy reported herein can be further expected to fabricate delivery systems of various water-soluble drugs.
1 Introduction
Given the advantages of high binding affinity, easy preparation and chemical stability, molecularly imprinted polymers (MIPs), as synthetic biomimetic receptors, have attracted considerable research interests from scientists and engineers due to their importance in many promising fields, such as tailor-made separation materials, chemical/biological sensors, artificial antibodies and drug delivery systems,1–4 etc. In spite of their wide range of uses, MIPs suffer from disadvantages including incomplete template removal, poor site accessibility to target species and the long diffusion paths required as a result of the bulk volume of the polymers.5 Addressing these problems, surface MIPs devices were developed, where the MIPs were coated on the surface or interface of various substrates by layer-by-layer (LbL) fashion,6 and underwent a rapid development thereafter.7 Meanwhile, some MIPs applications in the biomedical field, in particular for drug release, were reported because MIPs could greatly increase the residence time of the drug within the body by reducing the rate at which the drug was released,8 and reduce the drug toxicity or side effects during application. To date, various therapeutic agents such as tetracycline,9 propranolol,10 thiamine hydrochloride,11 timolol,12 norfloxacin,13 sulfasalazine,14 5-fluorouracil,15 methotrexate,16 ketotifen fumarate,17 copper salicylate,18 naltrexone,19 flufenamic acid,20 dipyridamole,8 and so on, have been released through MIPs approach.
In all the above-mentioned delivery systems, on the one hand, only a few examples have advanced to the practical and biomedical applications, mostly owing to their intrinsic toxicity (i.e., toxicity of monomers, cross-linker and initiator residues) and nondegradability. To resolve the issues associated with toxicity and nondegradability of the delivery carriers, some natural materials such as chitosan, alginate and genipin, are often employed as the polymeric segments to prepare the most promising MIPs because of their abundance, nontoxicity, biodegradability and biocompatibility properties. Chitosan which is a cationic mucopolysaccharide with similar structural characteristics to cellulose, as a low toxicity scaffold,21–24 has been one of the most popular biopolymers for development of drug delivery systems.25,26 In contrast, alginate is an anionic polysaccharide distributed widely in the cell walls of brown algae. Genipin derived from fruits of Gardenia jasminoides Ellis and Genipa americana, as a new natural cross-linker, has been successfully used in the engineering of molecularly imprinted chitosan hydrogels for recognizing o-xylene,27 and reported 5000 to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times less cytotoxic than glutaraldehyde.28,29 The use of natural crosslinkers to prepare polymers has become a promising alternative to fabricate fully biocompatible materials.30–32 On the other hand, the drug loading amount of MIPs is usually low (i.e., the loading amount of theophylline-MIPs was in the range of 0.1–2.0 mg g−1).33 For overcoming the problem, many recent studies have focused on the development of “hollow” capsules,34–36 which are extensively utilized in drug delivery systems due to their unique feature such as high loading capacity. But the drug release from the capsules is conventionally dependent on drug diffusion or degradation of networks. Furthermore, the release profile is insensitive to the changes of biological environment.37 Therefore, the exploitation of sophisticated delivery (release drug in a feedback-controlled manner) of active therapeutics from the capsules still remains a greatest challenge in the development of advanced drug delivery systems.
000 times less cytotoxic than glutaraldehyde.28,29 The use of natural crosslinkers to prepare polymers has become a promising alternative to fabricate fully biocompatible materials.30–32 On the other hand, the drug loading amount of MIPs is usually low (i.e., the loading amount of theophylline-MIPs was in the range of 0.1–2.0 mg g−1).33 For overcoming the problem, many recent studies have focused on the development of “hollow” capsules,34–36 which are extensively utilized in drug delivery systems due to their unique feature such as high loading capacity. But the drug release from the capsules is conventionally dependent on drug diffusion or degradation of networks. Furthermore, the release profile is insensitive to the changes of biological environment.37 Therefore, the exploitation of sophisticated delivery (release drug in a feedback-controlled manner) of active therapeutics from the capsules still remains a greatest challenge in the development of advanced drug delivery systems.
To address this challenge, we postulate that functionalized polymer microcapsules will make a good candidate imprinting materials having high-capacity uptake of target species through the complete removal of the template molecules, excellent site accessibility and hollow interior surface, that is to say, the microcapsules have a structure consisting of a hollow interior surrounded by a multilayer polysaccharide and thin imprinted shell. Furthermore, we presume that if the shell is sufficiently thin, a specific template drug of interest can be encapsulated inside microcapsule and in proximity of imprinted microcapsules' surface, in which the imprinted site will act as the gate or channel and the template as the stopper.38 The binding or release of drugs leads to the closing/opening of the gate and thus controls the transfusion of drugs through the mesochannels of imprinted microcapsules.
For this purpose, a three-step process was developed to form LbL polyelectrolyte microcapsules. First, we synthesized CaCO3 particles, as decomposable cores, whilst carboxylmethyl cellulose (CMC), as preincorporated polyanion, was coprecipitated at the moment of the CaCO3 core formation. Second, chitosan and alginate were used as the multilayer wall materials. After deposition of the multilayered materials, the cores were dissolved by Ca2+ complexation with ethylenediaminetetraacetic acid disodium salt (EDTA–2Na) to obtain hollow multilayer microcapsules, followed by loading of the model water-soluble drug, doxorubicin (DOX), which is a chemotherapy agent used in the treatment of several types of cancer. Finally, we selected O-carboxymethyl chitosan (O-CMCS) and genipin as functional monomer and cross-linking agent, respectively, to prepare the imprinted layers which were coated on the surface of hollow multilayer microcapsules (Fig. 1). Furthermore, DOX loading of the molecularly imprinted microcapsules (MIMs), pH-responsive drug release of DOX-loaded MIMs, cytotoxicity of MIMs against U373 cells as well as in vitro antitumor test of DOX-loaded MIMs were also performed.
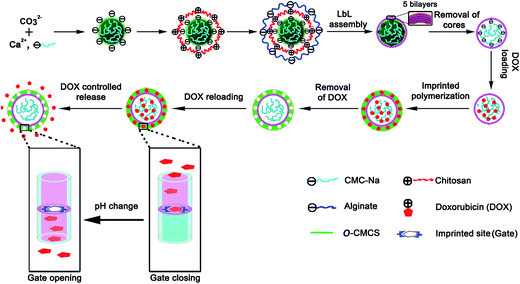 |
| | Fig. 1 The fabrication and formation mechanism of DOX-MIMs. | |
2 Experimental
2.1 Chemicals and materials
Chitosan (deacetylation degree 95% and molecular weight 40 kDa) was purchased from Jinan Haidebei Marine Bioengineering Co. Ltd. (Jinan, China). Sodium alginate (viscosity 250 cps) was purchased from Sigma-Aldrich (St. Louis, MO, USA). Carboxymethyl cellulose (CMC) sodium salt (viscosity 30–80 cps) was purchased from China Curative and Medicine Corporation (Shanghai, China). Genipin was purchased from Chengdu Kangbang Biotechnology Corporation (Chengdu, China) and doxorubicin (DOX) hydrochloride was purchased from Zhejiang Haizheng Pharmaceutics Corporation (Taizhou, Zhejiang, China). The water used in all experiments was triple distilled. All other chemicals and solvents were of analytical grade.
Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS) and antibiotics were purchased from Life Technologies Corporation (Gaithersburg, MD, USA). WST-1 cell proliferation and cytotoxicity assay kit were obtained from Beyotime Institute of Biotechnology (Haimen, Jiangsu, China).
U373 human malignant glioma cells were obtained from the American Type Culture Collection (Manassas, VA, USA) and cultured in DMEM supplemented with 10% FBS and 1% (v/v) antibiotics in a humidified incubator at 37 °C with 5% CO2.
2.2 Apparatus and analytical methods
The morphologies and structures of the DOX-MIMs were taken by an FEI Sirion-200 field emission scanning electron microscope (FE-SEM) from FEI (Hillsboro, OR, USA) and a JEM-2100 transmission electron microscopy (TEM) from JEOL (Tokyo, Japan). Bruker Vector 27 FT-IR spectrometer from Bruker (Ettlingen, Germany) was used to characterize the structure of O-CMCS. The amount of released or loaded DOX was measured by UV-2450 UV-vis spectrophotometer from Shimadzu (Kyoto, Japan). The cells were cultured in Heracell 150i CO2 incubator from Thermo Scientific Inc. (Waltham, MA, USA), and the absorbance of WST-1 assay was determined by Multiskan Microplate Reader from Thermo Scientific Inc. (Waltham, MA, USA). The size distributions of CaCO3(CMC) microparticles were measured by a Winner 2000 particle sizer from Jinan Winner Particle Technology Co. Ltd. (Jinan, China). Zeta potential of the CaCO3(CMC) microparticles coated with chitosan–alginate multilayers was measured in 0.2 M NaCl solution (pH 5.0) by a Zeta-sizer (Mastersizer 2000) from Malvern Instruments Ltd. (Malvern, UK). Each datum was averaged from three measurements.
2.3 Preparation of CaCO3 microparticles integrated with CMC
CaCO3 microparticles doped with CMC, abbreviated as CaCO3(CMC), were synthesized by mineralization of Ca(NO3)2 and Na2CO3 solutions in the existence of CMC.39 Briefly, 100 mL of 0.025 M Ca(NO3)2 solution was mixed with 2 mL of 5% CMC solution, into which 100 mL of 0.025 M Na2CO3 solution was rapidly poured under vigorous agitation for 30 s. After 30 min, the precipitated particles were collected by centrifugation and washed with triple-distilled water, and then dried under vacuum. Finally, the CaCO3(CMC) hybrid microparticles were obtained.
2.4 Fabrication of chitosan–alginate multilayer microcapsules (CAMMs)
The CAMMs were fabricated by a template-assisted assembly in a LbL fashion, followed by removal of cores.40 Solutions of chitosan (0.5 mg mL−1, pH 5.0) and alginate (1.0 mg mL−1, pH 5.0) were obtained by dissolving chitosan and alginate in 0.5 M NaCl solution, respectively. The CaCO3(CMC) hybrid microparticles were washed with 0.5 M NaCl solution (pH 5.0) for 3 times before the fabrication. In a typical fabrication process, the CaCO3(CMC) hybrid microparticles were incubated in the chitosan or alginate solution for 15 min. The multilayers were deposited onto the CaCO3(CMC) hybrid microparticles by consecutive adsorption of chitosan and alginate. Then the microparticles were separated by centrifuge and washed with 0.5 M NaCl solution (pH 5.0) for 3 times at each interval. The LbL procedure was repeated until 5 bilayers of polysaccharides were assembled with chitosan as the outermost layer. After the core–shell microparticles were cross-linked by genipin (2.5 mg mL−1) at 37 °C for 6 h, the CaCO3 particles were dissolved by 0.2 M EDTA–2Na solution for three times (each for 15 min), which gave rise to hollow CAMMs. The hollow CAMMs were washed three times with triple-distilled water before use.
2.5 Synthesis of O-CMCS
The O-CMCS was prepared from chitosan with minor modifications.41 In brief, chitosan (20 g), sodium hydroxide (27 g) and isopropanol (200 mL) were added into a flask to swell and alkalize at −20 °C for 24 h. Subsequently, monochloroacetic acid (30 g) was dissolved in isopropanol (40 mL), then added into the above mixture dropwise for 30 min and reacted at 50 °C for 7 h. And the reaction was stopped by adding 70% ethanol (400 mL) to the mixture. The solid was separated by filtration, rinsed by 70% ethanol for five times, dehydrated with absolute alcohol and dried under vacuum at room temperature. The obtained primary product was the sodium salt of O-CMCS. The sodium salt of O-CMCS (1 g) was converted to O-CMCS by immersing in 70% ethanol (100 mL) and adding 32% HCl (10 mL). Then the resultant suspension was stirred for 30 min and filtered. The final product, which was the H-form of O-CMCS, was rinsed in deionized, distilled water, dialyzed for three days to remove impurity and freeze-dried.
2.6 Preparation of hollow DOX-MIMs
Prior to the imprinted polymerization reaction, CAMMs (100 mg) were incubated in 10 mL of DOX solution (1.0 mg mL−1) at room temperature. After the adsorption equilibrium was reached, the DOX concentration of supernatant was consecutively adjusted until it was 1.0 mg mL−1. Meanwhile, O-CMCS (16 mg) and genipin (31 mg) were averagely divided into 5 parts and each part was added into above mixture under gentle stirring, each interval for 1 h, respectively. The reaction was performed under shaking at 37 °C for 12 h. The obtained microcapsules were cleaned by 1% (v/v) acetic acid solution to remove DOX, then washed with deionized, distilled water for several times and dried in vacuum oven at room temperature. The hollow non-imprinted microcapsules (NIMs) were also prepared under the same conditions but without the addition of the DOX template.
2.7 DOX loading
Hollow DOX-MIMs (20 mg), NIMs (20 mg) and CAMMs (20 mg) was suspended in 2.0 mL of normal saline (0.9% NaCl) with a feeding DOX concentration ranging from 0.17 to 10 mM at room temperature, respectively. After incubation under a reciprocating shaking-table for 12 h, the microcapsules in the solution were filtered through a 0.22 μm microporous membrane, and the DOX concentration of filtrate was measured using a UV-vis spectrophotometer at 253 nm. The loading amount of DOX was determined by measuring the difference between the total amount and the residual amount in solution. Each experiment was replicated at least three times.
2.8 DOX release
DOX-loaded MIMs (20 mg), DOX-loaded NIMs (20 mg) and DOX-loaded CAMMs (20 mg) was mixed in 2 mL of normal saline (pH = 6.5 or 5.0) and incubated at 37 °C under gentle shaking, respectively. At different time, a 10 μL aliquot of supernatant was removed and then supplemented with 10 μL fresh normal saline. Subsequently, the supernatant was diluted to desired concentration. The absorption at 253 nm was recorded. The cumulative release amount (%) of DOX was integrated from each measurement. Each experiment was replicated at least three times.
2.9 Cytotoxicity assay
Dry sample of hollow DOX-MIMs, NIMs and CAMMs was sterilized by UV exposure for 2 h and then dispersed in the cell culture medium with a final concentration of 1.0 mg mL−1, respectively.
The cytotoxicities of three samples on U373 cells were evaluated by WST-1 assay. Briefly, U373 cells were seeded into 96-well plates at a density of 5 × 103 cells per well in 100 μL DMEM medium containing 10% FBS and cultured for 24 h. The medium was then changed to fresh medium containing 0–100 μg mL−1 of DOX-MIMs, NIMs and CAMMs for another 48 h, respectively. After washing twice with phosphate buffer solution (PBS), 100 μL of medium containing 10 μL of WST-1 reagent was added to each well and incubated for another 4 h, and then the absorbance was read at 450 nm. Cell viability in each group was expressed as the percentage of the metabolic activity of treated cells with respect to untreated cells. Experiments were performed three times in quintuplicate.
2.10 In vitro antitumor test
Samples of DOX-loaded MIMs, DOX-loaded NIMs and DOX-loaded CAMMs were also prepared as described above. For the in vitro antitumor activity assay, U373 cells (5 × 103 cells per well) were seeded into 96-well plates and cultured overnight. Subsequently, the culture medium was changed to 150 μL of fresh medium containing a specific concentration of DOX (IC50, 7.5 μg mL−1), DOX-loaded MIMs (83.4 μg mL−1), DOX-loaded NIMs (84.4 μg mL−1) and DOX-loaded CAMMs (77.0 μg mL−1), respectively. Cells cultured in the blank medium were used as the negative control. After incubation for 24, 48, 72, 96, 120 and 144 h, cell viability was measured as described previously. Cell growth inhibition rate was calculated using the following equation:
| Inhibition rate % = (Acontrol − Asample)/Acontrol × 100% |
where Asample and Acontrol was denoted as absorbance of the cells treated with formulations and the blank culture medium, respectively.
3 Results and discussion
3.1 Synthesis and characterization of CaCO3(CMC) hybrid microparticles
Calcium carbonate has been of considerable interest due to its wide applications in medicine, industry and many other fields. To obtain the nonaggregated CaCO3 microparticles with uniform and homogeneous size in the synthesis process, CMC, a negatively charged polysaccharide, was added into the reaction system to control the macroscopic shape of the resultant CaCO3 microparticles. This endows also the derived CMC-containing microcapsules with a feature of spontaneous loading of positive charge drugs (such as DOX) by electrostatic interaction. SEM image (Fig. 2A) shows that the CaCO3(CMC) microparticles have the regular spherical shape and compact structure. The size distribution of the hybrid microparticles in aqueous solution measured by particle size analyzer is presented in Fig. 2B, the microparticles exhibit unimodal size distribution with relatively narrow distribution, and the size of the microparticles measured by particle size analyzer was 7.7 ± 0.8 μm. Compared with the size of water-soaked microparticles measured by particle size analyzer, the size of dried microparticles from SEM observation is smaller, resulting from the fact that the surface layer of microparticles shrink after the water is removed.
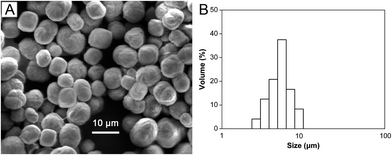 |
| | Fig. 2 SEM image (A) and size distribution (B) of CaCO3(CMC) hybrid microparticles. | |
3.2 Fabrication and characterization of hollow CAMMs
To increase the loaded DOX amount of the microcapsules and decrease release rate of the loaded DOX from the microcapsules, the CaCO3(CMC) hybrid microparticles were further coated with chitosan–alginate multilayers before removal of cores. The LbL self-assembly process was monitored by zeta potential as shown in Fig. 3A. Since CMC is negatively charged, the zeta potential of the CaCO3(CMC) microparticles was −23.33 ± 0.38 mV. Compared with the bare microparticles, the zeta potential of the microparticles deposited with the first chitosan layer still remained a negative value, but increased about 6 mV. After the second cycle of the assembly process, regular charge reversal of zeta potential indicated adsorption of polycation chitosan and polyanion alginate on the CaCO3(CMC) microparticles alternatingly. After 5 bilayers of chitosan–alginate were assembled, the surface of microparticles with chitosan as the outermost layer became rougher (Fig. 3B), which was distinctive with the pristine CaCO3(CMC) microparticles (Fig. 2). This can be explained by the formation of a polyelectrolyte net onto the microparticles' surface, which shrinks when dried for SEM measurements. Subsequently, the core–shell microparticles with chitosan outer layer were cross-linked by genipin. Previous researches have demonstrated that genipin reacts with materials containing primary amine groups (i.e., chitosan, some peptides and polypeptides) to form covalently cross-linked polymers.28,30,42–44 The crosslinks are formed by two reactions involving different sites on the genipin molecule (Fig. 4). One of the crosslinks is an SN2 reaction, that is to say, the ester group on the genipin molecule is replaced by a secondary amide linkage to form polymerized macromolecule (Fig. 4-a). When the ester substitution occurred, the other half of the crosslink, which is the ring opening reaction induced by nucleophilic attack, must have already formed. The reaction begins with an initial nucleophilic attack of the genipin C3 atom from a primary amine group of chitosan to form an intermediate aldehyde group, resulting in opening of the dihydropyran ring and producing the iridoid containing nitrogen. Then the iridoid makes an intermediate heterocyclic compound via dehydration and the intermediate generates the chromatic chitosan–genipin conjugates by polymerization (Fig. 4-b), the resultant cross-linked chitosan layer could be visualized. Interestingly, an apparent color change of the core–shell microparticles with different bilayers was clearly observed (Fig. 5). The more the amount of bilayers deposited on microparticles is, the deeper the color of the microparticles gets.
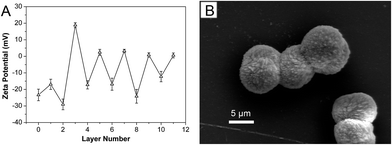 |
| | Fig. 3 (A) Zeta potential of CaCO3(CMC) microparticles coated with chitosan–alginate multilayer as a function of layer number (the odd and even numbers represent chitosan and alginate, respectively, except for number 0, bare microparticles). (B) SEM image of CaCO3(CMC)–(chitosan/alginate)5 microparticles. | |
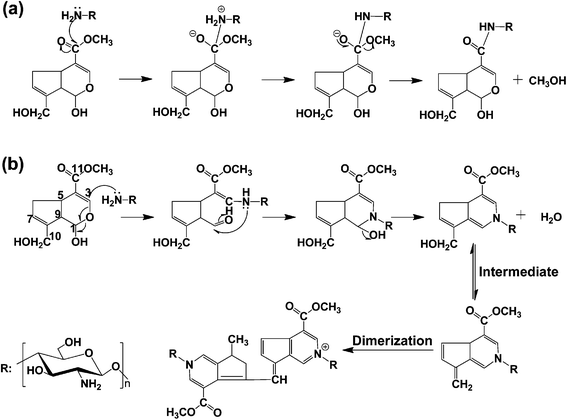 |
| | Fig. 4 The process of genipin cross-linked chitosan: (a) nucleophilic attack and (b) nucleophilic substitution. | |
 |
| | Fig. 5 The gray level transformation of core–shell microparticles with different bilayers: (a) 1 bilayer, (b) 2 bilayers, (c) 3 bilayers, (d) 4 bilayers and (e) 5 bilayers. | |
Finally, removal of the template particles by use of 0.2 M EDTA–2Na solution yielded hollow CAMMs. As shown in Fig. 6, together with the creases and folds that were typical features for thin microcapsules in the dry state, the complete removal of the template CaCO3 cores could be concluded. And the surface shape of the CAMMs cross-linked by genipin was smooth and glossy. All these results further confirmed the successful coating of the CaCO3(CMC) microparticles with the chitosan–alginate multilayers.
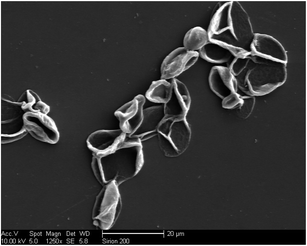 |
| | Fig. 6 SEM image of hollow CAMMs. | |
3.3 Synthesis and characterization of O-CMCS
The choice of the suitable monomer is a crucial parameter in the preparation of MIPs. According to the property of DOX molecule with positive charge, a candidate functional monomer with negative charge must be considered. O-CMCS is an amphiprotic ether derivative, which contains carboxy group and amino group in the molecule, and has many outstanding properties including water solubility, non-toxicity, biodegradability and biocompatibility. As a versatile biomedical material, O-CMCS always conjugates or entraps or self-assembles with agent and is safe in clinical application.41,45 For these advantages, O-CMCS was selected as a suitable functional monomer in this work. The O-CMCS could be synthesized by reacting chitosan with monochloroacetic acid in isopropanol as a solvent using the condensation reaction.
The chemical structure of original chitosan and product were confirmed by IR spectra (Fig. 7). Fig. 7-a shows the basic characteristics of chitosan at: 1090 cm−1 (C–O stretch), 1150 cm−1 (bridge-O stretch), 1554 cm−1 (N–H bend of primary amine), 2880 cm−1 (C–H stretch) and 3420 cm−1 (O–H stretch). Compared with infrared spectra of original chitosan, the sodium salt of O-CMCS displays the new absorption peaks at 1410 cm−1 and 1601 cm−1, corresponding to the symmetry and asymmetry stretch vibration of COO− (Fig. 7-b), respectively. Thus, the results of IR spectroscopy confirmed carboxymethylation of chitosan.
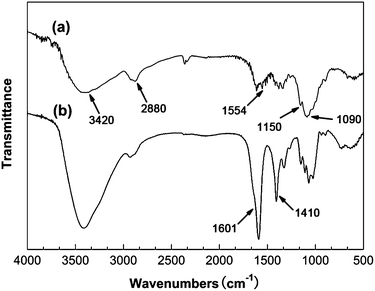 |
| | Fig. 7 FT-IR spectrum of chitosan (a) and sodium salt of O-CMCS (b). | |
3.4 Preparation and characterization of hollow DOX-MIMs
The target antitumor drug, DOX, was used as the template compound for the synthesis of MIMs, which controlled the release rate of DOX from the microcapsules. O-CMCS was selected as the functional monomer of the DOX binding event, because it contains primary amine groups necessary for the copolymerization to synthesize the MIMs. Genipin was employed as cross-linking agent, which could react with materials containing primary amine groups (chitosan and O-CMCS) to form covalently cross-linked networks. The molecular imprinting system was accomplished by direct copolymerization and cross-linking reactions in the presence of functional monomer (O-CMCS), cross-linking agent (genipin) and template molecule (DOX). The final Hollow DOX-MIMs were obtained after the templates were removed from the polymer shells by repeated extractions in 1% (v/v) acetic acid solution. Fig. 8 illustrates the monomer–template complex (Fig. 8-1), molecular imprinting polymerization (Fig. 8-2) and the extraction and rebinding of template molecules of the DOX-MIMs (Fig. 8-3). The copolymerization of chitosan outer layer with functional groups (–NH2) resulted in selective polymerization at the surface of CAMMs, and hence the formation of uniform shells. During the imprinting polymerization and cross-linking process, DOX in the form of complex was embedded into the cross-linked polymer matrix, as shown in Fig. 8-2. The template molecules could be extracted to form DOX-imprinted sites in the polymer network matrix when they were protonated in acidified solution, generating carboxyl groups anchored at the wall of DOX-imprinted cavities, as indicated with stuffed arrow in Fig. 8. So the resultant imprinted sites were complementary to the shape, size and chemical functionality of DOX. Under appropriate conditions, DOX could rebind to these cavities through electrostatic interaction with the carboxyl groups of O-CMCS.
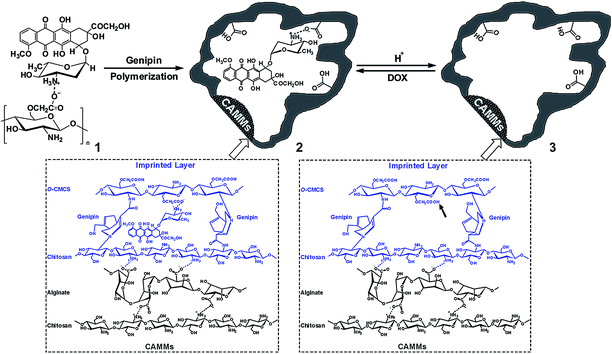 |
| | Fig. 8 Schematic representation of molecular imprinting of DOX at the surface of CAMMs using O-CMCS as functional monomers and genipin as cross-linking agent. | |
The hollow structure of the DOX-MIMs or NIMs was confirmed by SEM. All the microcapsules shows collapsed structure as a result of water evaporation during drying as shown in Fig. 9. Compared with the hollow CAMMs (Fig. 6), the shell thickness of hollow DOX-MIMs or NIMs was slightly thicker. The thickness of imprinted layer was about 20 nm, as calculated by measuring the difference of microcapsules' shell thickness after and before imprinting. Unfortunately, it is difficult to discriminate a thin imprinted shell and CAMMs shell by TEM because the two shells consist of same materials (polyelectrolytes). Indeed, we had performed a control experiment by synthesizing DOX-MIMs with thicker shells in order to validate the synthesis protocol of the core–shell microcapsules. But the thick-shell microcapsules are not suitable for gate-controlled drug release investigations, though, for which microcapsules with thin MIPs shells were always used. These findings further confirmed that the imprinted layers were successfully coated onto the hollow microparticles with the chitosan–alginate multilayers.
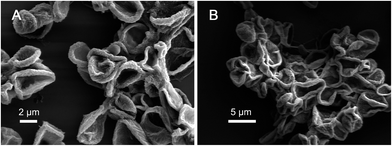 |
| | Fig. 9 SEM images of DOX-MIMs (A) and NIMs (B). | |
3.5 DOX loading
DOX is physically trapped inside the CMC cores of DOX-MIMs by simple electro-adsorption. Fig. 10A presents the loading capacities of the CAMMs, DOX-MIMs and NIMs. It was investigated with a series of DOX standard solutions of 0.17–10 mM in normal saline. The experiments revealed that the loading capacity continuously increased along with the increase of concentration in range of 0.17–10 mM for DOX-MIMs, DOX-NIMs and CAMMs and reached equilibrium as the concentration was up to 8.5 mM, respectively. The maximum loading capacity of the DOX-MIMs, DOX-NIMs and CAMMs was 155.1 μmol g−1, 153.3 μmol g−1 and 167.9 μmol g−1 for DOX, respectively. The data show clearly that there is not much difference in loading capacity between the three types of microcapsules. The loading capacity here is ascribed to the previously incorporated CMC, which possess the driving force for accumulation of the positively charged species. For further comparison, the loading capacity of DOX-MIMs or NIMs was even less than that of CAMMs because of having a smaller number of microcapsules per unit of weight, and when DOX concentration was close to equilibrium concentration (that is say, DOX concentration is in range of 5.0–9.0 mM), the loading capacity of DOX-MIMs was slightly more than that of DOX-NIMs due to the existence of the imprinted cavities of DOX-MIMs (the inset of Fig. 10A).
 |
| | Fig. 10 (A) The loading capacity of CAMMs (▼), DOX-MIMs (▲) and NIMs (●) to DOX, respectively (the inset indicates the loading capacities of three microcapsules to DOX after 5 mM). (B) TEM images of DOX-MIMs before (a) and after (b) DOX loading. | |
Besides the quantitative assay, the successful loading of DOX and its distribution within the microcapsules had also been directly confirmed by TEM (Fig. 10B). As observed under TEM, the hollow DOX-MIMs were collapsed (Fig. 10B-a). The creases and folds were typical features for ultrathin microcapsules in the dry state, implying the complete removal of the template CaCO3 cores. But this would not mean that the microcapsules were absolutely “hollow”, because the CMC macromolecules were partly encapsulated within the microcapsules by complexation with the excess chitosan adsorbed into the porous CaCO3 microparticles. Compared with the hollow DOX-MIMs (Fig. 10B-a), the dark shadow was observed in the center of the DOX-loaded MIMs with 8.2 ± 0.9 μm (Fig. 10B-b), demonstrating again the successful DOX loading.
3.6 DOX release
It is well known that the local pH of tumor tissue is lower than that of normal human tissue, i.e., the pH of normal tissue is about 7.4 and the pH of tumor tissue may be as low as 5 or 6.39 Here, the release behavior of the DOX-loaded microcapsules was investigated in normal saline with pH values of 6.5 and 5.0, respectively. The release profiles were shown in Fig. 11. At pH 6.5, DOX from DOX-loaded CAMMs or NIMs had significantly faster release rate than that from DOX-loaded MIMs. For example, DOX release amount of DOX-loaded MIMs was only ∼18% in 12 h, but DOX release amount of DOX-loaded NIMs or CAMMs reached up to ∼50% within the same period. Importantly, ∼100% of DOX was released from DOX-loaded MIMs in 168 h, and ∼100% of DOX was released from DOX-loaded CAMMs or NIMs in 72 h (Fig. 11A). The phenomenon indicated that the release rate of DOX-loaded MIMs was relatively slow after the initial burst release within the first 12 h, and the release process could be sustained to more than 168 h. This significant difference in the release of the MIMs and NIMs revealed the existence of specific sites in MIMs that interacted strongly with DOX molecules, so that a very long time was required for the acid solution to overcome the electrostatic interaction of DOX molecules with their specific sites. Obviously, DOX-loaded MIMs can realize the orderly release of DOX by responding to slightly acidic condition. The advantages of the controlled and delayed release of MIMs are increasing the residence time of drug within body, ensuring better compliance with most dosage requirements, and maintaining the drug concentration below the levels at which potential harmful side effects become prevalent (for the drug with a narrow therapeutic index). When the pH decreased to 5.0 (Fig. 11B), the DOX releases from three microcapsules were dramatically accelerated. Especially, DOX had slightly faster release rate from DOX-loaded MIMs than that at pH 6.5, that is to say, at pH 5.0, the proton substitution rate accelerated, leading to destruction of the electrostatic interactions between DOX and O-CMCS and the separation of DOX and imprinted sites. As a result, the drug was released more quickly. More than 50% of the drug was burst released from the three microcapsules within 8 h, and ∼75% of the drug was released in 24 h.
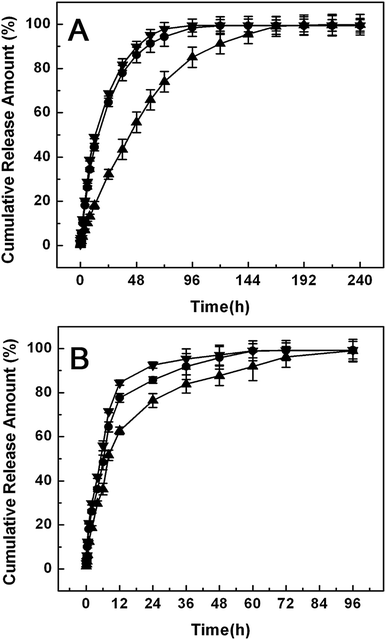 |
| | Fig. 11 The release profiles of CAMMs (▼), DOX-MIMs (▲) and NIMs (●) loaded with DOX at different pH: (A) pH 6.5 and (B) pH 5.0. | |
3.7 Cytotoxicity of DOX-MIMs against U373 cells
To assess the biocompatibility of the plain polyelectrolyte microcapsules, we performed the cytotoxicity assays of pure DOX-MIMs, NIMs and CAMMs against U373 cells. Compared with the morphology of U373 cells cultured in blank medium (Fig. 12A-a), that of U373 cells cultured in CAMMs, DOX-MIMs and NIMs has no obvious difference under microscopic inspection, respectively (as indicated with hollowed arrow in Fig. 12A-b, c and d). WST-1 assay results show that the absorbance of U373 cells also had no significant change after contacted with different concentrations of CAMMs, DOX-MIMs and NIMs (0 to 100 μg mL−1) for 48 h (Fig. 12B). The cell viability, which was normalized to that of U373 cells cultured with blank culture medium (treated with 0 μg mL−1 microcapsules), maintained about 100% after incubated with 20–100 μg mL−1 of DOX-MIMs, NIMs and CAMMs, respectively (Fig. 12C). These results indicated that the DOX-MIMs had a good biocompatibility.
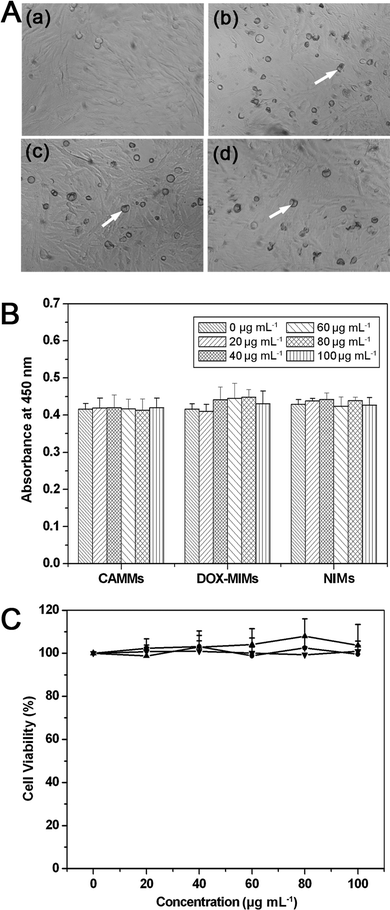 |
| | Fig. 12 Cytotoxicity of the three types of microcapsules against U373 cells. (A) Microscopic photographs of U373 cells cultured in blank medium (a), 100 μg mL−1 CAMMs (b), 100 μg mL−1 DOX-MIMs (c) and 100 μg mL−1 NIMs (d), respectively. Magnification, ×100. (B) WST-1 assay results (absorbances) of U373 cells in contact with different concentrations (0–100 μg mL−1) of CAMMs (▼), DOX-MIMs (▲) and NIMs (●) for 48 h. (C) Cell viability based on the results of WST-1 assay. Data are means ± SD for three separate experiments. | |
3.8 In vitro antitumor test
In order to evaluate the anti-cancer potential of DOX-loaded MIMs, we performed a serious of in vitro cell growth inhibition assays on U373 cells using free DOX as the positive control. The cells were exposed to a specific concentration of DOX-loaded microcapsules (MIMs, NIMs and CAMMs). Here, the amount of the free DOX was equal to the amount of DOX loaded into each type of microcapsules, that is to say, a final DOX concentration of DOX-loaded MIMs, NIMs and CAMMs in the culture medium was ∼7.5 μg mL−1, respectively. As shown in Table 1, all formulations, including DOX-loaded microcapsules (MIMs, NIMs and CAMMs) and free DOX, exhibited the inhibitory effects on cell growth. The cell inhibition rate of DOX-loaded MIMs group was similar to that of free DOX treated group at 48 h (48.11% vs. 49.79%), 96 h (64.22% vs. 67.41%) and 144 h (70.14% vs. 76.10%), which demonstrated the bioequivalence of 83.4 μg mL−1 DOX-loaded MIMs with 7.5 μg mL−1 free DOX. By close inspecting Table 1, it could be seen that the inhibition rate of each DOX-loaded MIMs group was higher than that of free DOX treated group in the first 24 h (similar results were also obtained for DOX-loaded NIMs and CAMMs groups). The result is just the reverse of what we expected. One main reason is that chitosan or O-CMCS as the outer layer of microcapsules itself has a certain antitumor activity and its positive charge can neutralize the negative charge on the tumor cell surface, resulting in selective absorption. So, the chitosan microcapsules can increase drug concentration in the tumor site and inhibition rate of tumor-cell.46 The other reason may be that chitosan or O-CMCS can act on tumor cells directly to interfere with cell metabolism, inhibit cell growth, or induce cell apoptosis.47 Interestingly, we also found that the inhibition rate of DOX-loaded MIMs group steadily increased at an average rate of 8 percent over a time period of 144 h, but the cell growth inhibition rate of DOX-loaded NIMs or CAMMs group increased in a nonlinear manner with a 24 h interval. The result further implies that DOX-loaded MIMs may be a good carrier for controlled-drug delivery to the tumor cell.
Table 1 Cell growth inhibition rate after treated with free DOX, DOX-loaded MIMs, DOX-loaded NIMs and DOX-loaded CAMMs, respectivelya
| Time (h) |
Cell growth inhibition rate (%) |
| DOX |
DOX-loaded MIMs |
DOX-loaded NIMs |
DOX-loaded CAMMs |
| Data are represented as mean ± SD. *P < 0.05, compared to cells treated with DOX-loaded MIMs for 24 h. |
| 24 |
31.81 ± 1.92* |
40.28 ± 1.61 |
37.88 ± 1.17 |
36.76 ± 0.65 |
| 48 |
49.79 ± 2.70 |
48.11 ± 1.36 |
38.74 ± 2.23 |
37.83 ± 1.65 |
| 72 |
52.70 ± 2.55 |
57.35 ± 1.15 |
51.65 ± 3.08 |
42.91 ± 0.37 |
| 96 |
67.41 ± 0.87 |
64.22 ± 1.17 |
55.87 ± 1.29 |
51.57 ± 2.28 |
| 120 |
67.86 ± 2.63 |
69.01 ± 0.46 |
59.89 ± 0.79 |
53.82 ± 2.36 |
| 144 |
70.10 ± 1.99 |
77.14 ± 0.66 |
66.33 ± 1.50 |
64.13 ± 1.11 |
4 Conclusion
In summary, we have successfully developed a hybrid strategy, which combines LbL assembly and molecular imprinting technique, for the synthesis of high-quality DOX-MIMs. The O-CMCS, acting as the functional monomer, are capable of complexing with DOX and copolymerizing with chitosan to afford imprinted cavities with binding sites. DOX can be loaded into MIMs while the further polyelectrolyte imprinting layer serves as the release controlling membrane. The resultant hollow DOX-MIMs showed the high loading capacity of hydrophilic anticancer drug DOX by simply soaking in DOX solution, which could be ascribed to the spontaneous deposition of DOX into the hollow polyelectrolyte microcapsules by the driving force of electrostatic interaction between DOX and polyelectrolyte microcapsules (CMC and O-CMCS). Upon exposure to a weakly acidic condition, the DOX-loaded MIMs released DOX to a greater extent through pH-response and gate-controlled manner, in contrast to burst release of DOX-loaded NIMs or CAMMs. The result suggests that DOX can transfer effectively into the exterior of the microcapsules via the imprinted sites, which can be blocked by the template molecules. The cytotoxicity of the DOX-loaded MIMs was also evaluated with U373 malignant glioma cells, which exhibited a better anti-cancer efficacy (than that of the free drug). Meanwhile, the hollow DOX-MIMs did not show any noticeable cytotoxicity, confirming the highly biocompatible and nontoxic features of polyelectrolyte imprinting microcapsules. Although the current work is mainly focused on the imprinting of DOX molecules, we have demonstrated that the imprinting technique at the surface of CAMMs is usually applicable to imprinting other aqueous soluble drugs such as 5-fluorouracil and pirarubicin. Based on above results, we are convinced that such polyelectrolyte imprinting microcapsules will be applied to the localized anticancer drug delivery, which increases drug accumulation in tumor cells and reduces adverse side effects on healthy organs by limiting drug concentration in the blood. Further investigation of in vivo DOX release tests using animal models are currently underway for evaluating its treatment effect on curing malignant glioma.
Acknowledgements
This work is supported by the National Natural Science Foundation of China (no. 21275075) and the Specialized Research Fund for the Doctoral Program of Higher Education of China (no. 20113234110001).
References
- M. Rejtharová and L. Rejthar, J. Chromatogr. A, 2009, 1216, 8246–8253 CrossRef PubMed.
- Y. X. Wang, Y. Ding, F. Rong and D. G. Fu, Polym. Bull., 2012, 68, 1255–1270 CrossRef CAS.
- D. Kriz, O. Ramstrom and K. Mosbach, Anal. Chem., 1997, 69, 345A–349A CrossRef CAS.
- K. Haupt and K. Mosbach, Trends Biotechnol., 1998, 16, 468–478 CrossRef CAS.
- D. M. Gao, Z. P. Zhang, M. H. Wu, C. G. Xie, G. J. Guan and D. P. Wang, J. Am. Chem. Soc., 2007, 129, 7859–7866 CrossRef CAS PubMed.
- J. Gauczinski, Z. Liu, X. Zhang and M. Schönhoff, Langmuir, 2010, 26, 10122–10128 CrossRef CAS PubMed.
- Y. Zhou, M. J. Cheng, X. Q. Zhu, Y. J. Zhang, Q. An and F. Shi, ACS Appl. Mater. Interfaces, 2013, 5, 8308–8313 CAS.
- M. Esfandyari-Manesh, M. Javanbakht, F. Atyabi, A. Mohammadi, S. Mohammadi, B. Akbari-Adergani and R. Dinarvand, Mater. Sci. Eng., C, 2011, 31, 1692–1699 CrossRef CAS PubMed.
- W. Cai and R. B. Gupta, Sep. Purif. Technol., 2004, 35, 215–221 CrossRef CAS.
- C. Bodhibukkana, T. Srichana, S. Kaewnopparat, N. Tangthong, P. Bouking, G. P. Martin and R. Suedee, J. Controlled Release, 2006, 113, 43–56 CrossRef CAS PubMed.
- T. S. Anirudhan, P. L. Divya and J. Nima, React. Funct. Polym., 2013, 73, 1144–1155 CrossRef CAS PubMed.
- H. Hiratani and C. A. Lorenzo, Biomaterials, 2004, 25, 1105–1113 CrossRef CAS.
- C. Alvarez-Lorenzo, F. Yanez, R. Barreiro-Iglesia and A. Concheiro, J. Controlled Release, 2006, 113, 236–244 CrossRef CAS PubMed.
- F. Puoci, E. Iemma, R. Muzzalupo, U. G. Spizzirri, S. Trombino, R. Cassano and N. Picci, Macromol. Biosci., 2004, 4, 22–26 CrossRef CAS PubMed.
- W. T. Kan and X. Li, Eur. Polym. J., 2013, 49, 4167–4175 CrossRef CAS PubMed.
- M. Curcio, O. I. Parisi, G. Cirillo, U. G. Spizzirri, F. Puoci, F. Iemma and N. Picci, E Polymer, 2009, 78, 1–7 Search PubMed.
- A. Tieppo, C. J. White, A. C. Paine, M. L. Voyles, M. K. McBride and M. E. Byrne, J. Controlled Release, 2012, 157, 391–397 CrossRef CAS PubMed.
- V. S. Sumi, R. Kala, R. S. Praveen and T. P. Rao, Int. J. Pharm., 2008, 349, 30–37 CrossRef CAS PubMed.
- K. Rostamizadeh, M. Vahedpour and S. Bozorgi, Int. J. Pharm., 2012, 424, 67–75 CrossRef CAS PubMed.
- M. S. da Silva, F. L. Nobrega, A. Aguiar-Ricardo, E. J. Cabrita and T. Casimiro, J. Supercrit. Fluids, 2011, 58, 150–157 CrossRef PubMed.
- D. Zhang, P. Sun, P. Li, A. B. Xue, X. K. Zhang, H. Y. Zhang and X. B. Jin, Biomaterials, 2013, 34, 10258–10266 CrossRef CAS PubMed.
- Y. Lu, D. L. Slomberg and M. H. Schoenfisch, Biomaterials, 2014, 35, 1716–1724 CrossRef CAS PubMed.
- M. Y. Wang, H. Y. Hu, Y. Q. Sun, L. P. Qiu, J. Zhang, G. N. Guan, X. L. Zhao, M. X. Qiao, L. Cheng, L. F. Cheng and D. W. Chen, Biomaterials, 2013, 34, 10120–10132 CrossRef CAS PubMed.
- J. J. Li, Y. Shu, T. Hao, Y. Wang, Y. F. Qian, C. M. Duan, H. Y. Sun, Q. X. Lin and C. Y. Wang, Biomaterials, 2013, 34, 9071–9081 CrossRef CAS PubMed.
- Y. C. Luo and Q. Wang, Int. J. Biol. Macromol., 2014, 64, 353–367 CrossRef CAS PubMed.
- R. Vivek, V. N. Babu, R. Thangam, K. S. Subramanian and S. Kannan, Colloids Surf., B, 2013, 111, 117–123 CrossRef CAS PubMed.
- B. M. Espinosa-Garcia, W. M. Argueelles-Monal, J. Hernandez, L. Felix-Valenzuela, N. Acosta and F. M. Goycoolea, Biomacromolecules, 2007, 8, 3355–3364 CrossRef CAS PubMed.
- H. W. Sung, R. N. Huang, L. L. Huang and C. C. Tsai, J. Biomater. Sci., Polym. Ed., 1999, 10, 63–78 CrossRef CAS PubMed.
- F. L. Mi, Y. C. Tan, H. C. Liang and H. W. Sung, Biomaterials, 2002, 23, 181–191 CrossRef CAS.
- F. L. Mi, H. W. Sung and S. S. Shyu, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2804–2814 CrossRef CAS.
- C. C. Tsai, R. N. Huang, H. W. Sung and C. L. Huang, J. Biomed. Mater. Res., 2000, 52, 58–65 CrossRef CAS.
- M. F. Butler, N. Yui-Fai and P. D. A. Pudney, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3941–3953 CrossRef CAS.
- M. C. Norell, H. S. Andersson and I. A. Nicholls, J. Mol. Recognit., 1998, 11, 98–102 CrossRef CAS.
- Q. H. Zhao, B. S. Han, Z. H. Wang, C. Y. Gao, C. H. Peng and J. C. Shen, Nanomedicine: Nanotechnology, Biology and Medicine, 2007, 3, 63–74 CrossRef CAS PubMed.
- M. L. De Temmerman, J. Demeester, F. De Vos and S. C. De Smedt, Biomacromolecules, 2011, 12, 1283–1289 CrossRef CAS PubMed.
- G. F. Luo, X. D. Xu, J. Zhang, J. Yang, Y. H. Gong, Q. Lei, H. Z. Jia, C. Li, R. X. Zhuo and X. Z. Zhang, ACS Appl. Mater. Interfaces, 2012, 4, 5317–5324 CAS.
- M. Dadsetan, K. E. Taylor, C. Yong, Z. Bajzer, L. Lu and M. J. Yaszemski, Acta Biomater., 2013, 9, 5438–5446 CrossRef CAS PubMed.
- C. D. Ki and J. Y. Chang, Macromolecules, 2006, 39, 3415–3419 CrossRef CAS.
- C. Y. Peng, Q. H. Zhao and C. Y. Gao, Colloids Surf., A, 2010, 353, 132–139 CrossRef CAS PubMed.
- Q. H. Zhao, Z. W. Mao, C. Y. Gao and J. C. Shen, J. Biomater. Sci., Polym. Ed., 2006, 17, 997–1014 CrossRef CAS PubMed.
- M. L. Zheng, B. Q. Han, Y. Yang and W. S. Liu, Carbohydr. Polym., 2011, 86, 231–238 CrossRef CAS PubMed.
- F. L. Mi, H. W. Sung and S. S. Shyu, J. Appl. Polym. Sci., 2001, 81, 1700–1711 CrossRef CAS.
- F. L. Mi, Y. C. Yan, H. C. Liang, R. N. Huang and H. W. Sung, J. Biomater. Sci., Polym. Ed., 2001, 12, 835–850 CrossRef CAS PubMed.
- F. L. Mi, H. W. Sung and S. S. Shyu, Carbohydr. Polym., 2002, 48, 61–72 CrossRef CAS.
- R. Jayakumar, M. Prabaharan, S. V. Nair, S. Tokura, H. Tamura and N. Selvamurugan, Prog. Mater. Sci., 2010, 55, 675–709 CrossRef CAS PubMed.
- J. J. Wang, Z. W. Zeng, R. Z. Xiao, T. Xie, G. L. Zhou, X. R. Zhan and S. L. Wang, Int. J. Nanomed., 2011, 6, 765–774 CAS.
- J. Cao and N. J. Zhou, Chin. J. Biochem. Pharm., 2005, 26, 127 Search PubMed.
Footnote |
| † These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here. 










