 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Well-defined ABA- and BAB-type block copolymers of PDMAEMA and PCL†
Carl Brucea,
Irakli Javakhishvilib,
Linda Fogelströma,
Anna Carlmarka,
Søren Hvilstedb and
Eva Malmström*a
aKTH Royal Institute of Technology, School of Chemical Science and Engineering, Department of Fibre and Polymer Technology, Teknikringen 56-58, SE-100 44 Stockholm, Sweden. E-mail: mavem@kth.se
bTechnical University of Denmark, Department of Chemical and Biochemical Engineering, Danish Polymer Centre, Building 227, DK-2800 Kgs Lyngby, Denmark
First published on 18th June 2014
Abstract
Triblock copolymers of ABA- and BAB-type consisting of poly(2-(dimethylamino)ethyl methacrylate) (PDMAEMA, A) and poly(ε-caprolactone) (PCL, B) have successfully been prepared. PDMAEMA-b-PCL-b-PDMAEMA (ABA) and PCL-b-PDMAEMA-b-PCL (BAB) were synthesised by a combination of ring-opening polymerisation of ε-CL, atom transfer radical polymerisation of DMAEMA and end-group conversion, performed through either acylation or azide–alkyne “click” chemistry. All samples were analysed by size exclusion chromatography where it was found that the evaluation of PDMAEMA-containing polymers was difficult due to the thermoresponsivity of PDMAEMA, affecting the solubility of the polymer in the temperature range at which the SEC was operated. From differential scanning calorimetry measurements it was shown that the crystallinity could be altered by changing the order of the blocks; with PDMAEMA as the outer block (ABA), the inherent crystallinity of PCL was destroyed while with PCL as the outer block (BAB), the degree of crystallinity was in the same proximity as for a PCL homopolymer.
Introduction
Polymer architecture is of great interest as it enables tailoring of the properties for a final application. A highly interesting architecture is well-defined block copolymers, consisting of two or more blocks, which can, for instance, be utilised as self-assembled structures or as interfacial bridging molecules. Atom Transfer Radical Polymerisation (ATRP)1 is one technique available for the synthesis of well-defined block copolymers of, for example, methacrylates,2–5 acrylates6–8 or styrenes.9–11 ATRP yields polymers with low molar-mass dispersity (ĐM), controlled molecular weight and preserved end-group functionality.12 Furthermore, when aiming for micellar applications, amphiphilic block copolymers, i.e., block copolymers composed of hydrophobic and hydrophilic segments are of particular interest.When an amphiphilic polymer is subjected to a solvent, solubilising only one of the segments, the chains of the insoluble block will self-assemble to form a micelle if the hydrophilic/hydrophobic balance is appropriate.13,14 Micelles have gained significant attention since they can be used to encapsulate and transport, and subsequently also release, compounds, such as in drug delivery.15–17 Micelles also have the ability to alter their functionality and/or the surface energy of a substrate through adsorption, an approach which has attracted increasing attention lately.18,19
For water-based applications, there are several options for the hydrophilic block as reported previously in literature, such as: poly(ethylene glycol) (PEG),20 poly(acrylic acid),21 and poly(N-isopropyl acrylamide) (PNIPAAm).22 One polymer that has gained attention due to its facile synthesis by ATRP, as well as due to the fact that it can carry a permanent cationic charge, is poly(2-(dimethylamino)ethyl methacrylate) (PDMAEMA). Several reports describe the ability of PDMAEMA to enable micellar formation in combination with a suitable hydrophobic block.23–28 However, PDMAEMA is prone to hydrolysis, wherefore its quaternised form, i.e., with a permanent charge, is preferred in water-based applications.29
In this study, triblock copolymers of PDMAEMA and poly(ε-caprolactone) (PCL) have been synthesised. The PDMAEMA block was synthesised through ATRP and the PCL block through Ring-Opening Polymerisation (ROP) of ε-caprolactone (ε-CL). ROP is a suitable method for polymerisation of cyclic lactones and, as ATRP, yields well-defined polymers of low ĐM.30 PCL is a biodegradable polymer previously used in a variety of applications such as medical31–33 and biocomposites.34–36 Block copolymers of PDMAEMA and PCL have previously been reported, mainly in the form of diblock copolymers, where different synthetic routes have been evaluated.37–39 However, the work on triblock copolymers of PDMAEMA and PCL is to a more limited extent reported in literature.40,41 Additionally, the reports on triblocks of PDMAEMA and PCL are, to the authors' best knowledge, limited to ABA type triblocks where PCL composes the middle block, i.e., B. In this work, the focus has been to synthesise well-defined ABA- and BAB-type triblocks of PDMAEMA and PCL, where PCL is B and PDMAEMA is A. In a long-term perspective it is interesting to evaluate both types of triblocks, ABA and BAB, as compatibilisers in cellulose-reinforced biocomposites.
Cellulose is the most abundant biopolymer in the world.42 In addition to being both biobased and biodegradable, cellulose fibres exhibit good mechanical properties, have a high aspect ratio and relatively low density.43 Cellulose fibres are therefore a good alternative to fossil-based fibres in composites. However, due to the hydrophilic nature of cellulose, the compatibility with many polymer matrices is poor and a modification is, therefore, desirable to perform in order to solve the compatibility issue.44,45 ABA and BAB triblocks would be two new types of compatibilisers for cellulose-reinforced composites. ABA triblocks can potentially form links between cellulose fibres or loops on one single fibre and BAB triblocks can theoretically form entanglements together with a polymeric matrix material and/or with other BAB or ABA block copolymers adsorbed. Hence, the development and optimisation of the synthesis of both types of triblocks are of interest. In this study the purpose is to, in a more fundamental approach, synthesise both types of triblocks for future evaluations in a final composite. The chemical approaches utilised herein were combinations of ATPR, ROP, end-group transformation through either acylation of a terminal hydroxyl group into an ATRP-initiator or of a terminal halide to an azide46 and Huisgen 1,3-dipolar cycloaddition reaction between alkynes and azides47 in its modified form, the Cu(I)-catalysed azide–alkyne “click” reaction.48
Experimental
Materials
All chemicals were purchased from Aldrich unless stated otherwise.2-(Dimethylamino)ethyl methacrylate (DMAEMA, 98%) was passed through a basic Al2O3 column prior to use to remove the inhibitor. ε-Caprolactone (ε-CL, 99%, Alfa Aesar) was dried over CaH2 overnight, distilled under reduced pressure, and stored under argon at 4 °C. Toluene (HPLC grade, Fischer Scientific) was dried through azeotropic boiling prior to use. 2-Hydroxyethyl bromoisobutyrate (HEBI, 95%), α-bromoisobutyryl bromide (BiB, 98%), 4-(dimethyloamino)pyridine (DMAP, 99+%), 1,1,4,7,10,10-hexamethyltriethylenetetramine (HMTETA, 97%), N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA, 99%), propargyl alcohol (99%), copper chloride (Cu(I)Cl, (99+%)), copper bromide (Cu(I)Br, (98%)), copper(II) bromide (Cu(II)Br2 (99%)), tin octoate (Sn(Oct)2, 95%), sodium azide (NaN3, 99+%), Sephadex G50 (medium), methyl iodide (MeI, 99%, Lancaster), sodium hydroxide pellets (NaOH, 98%), trimethylsilyldiazomethane solution (2 M in diethyl ether), hydrochloric acid (HCl, 37%), acetone (Prolabo, technical), methanol (MeOH, Merck), N,N-dimethylformamide (DMF, VWR), tetrahydrofuran (THF, Merck), and dichloromethane (DCM, Merck) were all used as received.
Methods
Nuclear magnetic resonance (NMR) spectra were recorded with a Bruker Avance 400 MHz using deuterated chloroform (CDCl3) as solvent. The residual solvent signal was used as the internal standard.To determine molecular weight (Mn), molar-mass dispersity (ĐM) and monitor ROP reactions, two size exclusion chromatography (SEC) systems were used: (1) TOSOH EcoSEC HLC-8320GPC system equipped with an EcoSEC RI detector and three columns (PSS PFG 5 μm; Microguard, 100 Å and 300 Å) (Mw resolving range: 300–100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da) from PSS GmbH utilising DMF (0.2 mL min−1) with 0.01 M LiBr as the mobile phase at 50 °C. A conventional calibration method was created using narrow molecular-weight distribution linear poly(methyl methacrylate) standards (Mw range: 400–300000 Da). The flow-rate fluctuations were corrected using toluene as an internal standard. PSS WinGPC software version 7.2 was used to process the data. (2) Viscotek 200 instrument using two PLgel mixed-D columns (Polymer Laboratories (PL)), assembled in series, and a refractive index detector, utilising THF (1 mL min−1) as the mobile phase operating at room temperature. Molecular weights were calculated using polystyrene (PS) standards from PL using TriSEC software.
000 Da) from PSS GmbH utilising DMF (0.2 mL min−1) with 0.01 M LiBr as the mobile phase at 50 °C. A conventional calibration method was created using narrow molecular-weight distribution linear poly(methyl methacrylate) standards (Mw range: 400–300000 Da). The flow-rate fluctuations were corrected using toluene as an internal standard. PSS WinGPC software version 7.2 was used to process the data. (2) Viscotek 200 instrument using two PLgel mixed-D columns (Polymer Laboratories (PL)), assembled in series, and a refractive index detector, utilising THF (1 mL min−1) as the mobile phase operating at room temperature. Molecular weights were calculated using polystyrene (PS) standards from PL using TriSEC software.
Fourier transform infrared (FTIR) spectra were recorded on a Perkin-Elmer Spectrum 100 FTIR equipped with a MKII Golden Gate™, Single Reflection ATR System from Specac Ltd, London, UK. The ATR-crystal was a MKII heated Diamond 45 °C ATR Top Plate.
The thermal properties of the polymers were analysed with differential scanning calorimetry (DSC). The experiments were performed with a Mettler-Toledo DSC with Mettler Toledo STARe software v9.2 equipped with a sample robot and a cryo-cooler. The heating and cooling rates were 10 °C min−1 in the temperature range of −70 °C to 180 °C. The degree of crystallinity (Xc) was calculated according to:
 | (1) |
 | (2) |
Dynamic light scattering (DLS) (Malvern Zetasizer NanoZS) was used to determine the hydrodynamic radius of the block copolymer in DMF with the addition of 0.01 M LiBr. The concentration of the solutions studied was 100 mg L−1.
Matrix-assisted laser desorption ionisation time-of-flight mass spectroscopy (MALDI-ToF MS) was conducted on a Bruker UltraFlex MALDI-ToF MS with a Scout-MTP Ion Source (Bruker Daltonics) equipped with a N2-laser (337 nm), a gridless ion source, and a reflector. All spectra were acquired by a reflector-positive method with an acceleration of 25 kV and a reflector voltage of 26.3 kV. The laser intensity was set to the lowest value possible to acquire high-resolution spectra. The instrument was calibrated with SpheriCal™ calibrants kindly supplied by Polymer Factory Sweden AB. THF solutions of 2,5-dihydroxybenzoic acid (DHB) and trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]malononitrile (DCTB) were prepared (10 mg mL−1) and used as matrices. The solution of DCTB was doped with sodium chloride. The dried-droplet sample-preparation technique, adapted from Cordeiro et al.,51 was used to obtain a 1:1 ratio (sample/matrix). 1 μL of each sample, PDMAEMA or PCL, was directly applied on the MALDI plate, followed by the addition of 1 μL of matrix solution, DHB for PDMAEMA and DCTB for PCL, and the sample was allowed to dry at room temperature to yield matrix crystallisation. The obtained spectra were analysed with FlexAnalysis Bruker Daltonics version 2.2.
Synthesis
All round bottom flasks and Schlenk tubes used in the reactions were equipped with stirring bars and closed with rubber septa, followed by drying with a heat gun – twice while evacuating and once under argon flow prior to use.ROP of ε-CL
All glass equipment was dried overnight prior to use in an oven at 150 °C in addition to the regular drying procedure. Sn(Oct)2 (250 mg, 0.63 mmol) was introduced into a two-necked flask and dissolved in THF (3.0 mL) under stirring for 30 min. HEBI (0.29 g, 1.5 mmol) was dissolved in THF (1.0 mL) in a glass vial. ε-CL (17 mL, 0.15 mol), the solution of HEBI and the solution of Sn(Oct)2 were injected into a Schlenk tube under a flow of argon, and stirred for 30 min at room temperature. The solution went from opaque to clear. The tube was then immersed in an oil bath preheated to 62 °C and the evolution of molecular weight was monitored by SEC in THF by withdrawing samples from the reaction mixture under argon flow. After 6 h the reaction mixture was allowed to cool down to ambient temperature, exposed to air, dissolved in THF and precipitated in a 10-fold excess of cold MeOH (−20 °C). The polymer was recovered via filtration and dried under reduced pressure at room temperature for 24 h. Yield: 6.0 g, 35%, calculations based on 100% conversion. The product was denoted Br-PCL-OH and characterised by SEC and NMR analysis. 1H NMR (400 MHz; CDCl3) δ (ppm): 4.22–4.35 (4H, m, BrC(CH3)2C(O)OCH2CH2OC(O)(CH2)5), 4.00 (2H, t, J = 5, –(CH2)4CH2OC(O)–), 3.58 (2H, t, J = 6.3, –(CH2)4CH2OH), 2.25 (2H, t, J = 6.3, –OC(O)CH2(CH2)4–), 1.87 (6H, s, Br(C(CH3)2C(O)O–)), 1.59 (4H, m, –OC(O)CH2CH2CH2CH2CH2–), 1.32 (2H, m, –OC(O)CH2CH2CH2CH2CH2).End-group conversion of Br-PCL-OH
DMAP (11 mg, 0.086 mmol) and TEA (11 mg, 1.0 mmol) were added to a two-necked round-bottomed flask, and dissolved in THF (5.0 mL) at room temperature for 10 minutes. The flask was then placed in an ice bath followed by slow addition of BiB (89 mg, 0.39 mmol), upon which the mixture went from transparent to white. After an additional 10 minutes, the solution of Br-PCL-OH (50 mg, 0.086 mmol) dissolved in THF (5.0 mL), was added drop-wise to the flask and the reaction was allowed to proceed overnight. The reaction mixture was precipitated twice in a 10-fold excess of cold MeOH (−20 °C) and recovered via filtration, followed by drying at reduced pressure at room temperature for 24 h. Yield: 0.35 mg, 68%, calculations based on 100% conversion. The product was denoted Br-PCL-Br, B, and characterised by NMR, SEC, MALDI-ToF and DSC analysis. 1H NMR (400 MHz; CDCl3) δ (ppm): 4.22–4.35 (4H, m, BrC(CH3)2C(O)OCH2CH2OC(O)(CH2)5O(O)C(CH3)2CBr), 4.00 (2H, t, J = 5, –(CH2)4CH2OC(O)–), 2.25 (2H, t, J = 6.3, –OC(O)CH2(CH2)4–), 1.87 (6H, s, Br(C(CH3)2C(O)O–)), 1.59 (4H, m, –OC(O)CH2CH2CH2CH2CH2–), 1.32 (2H, m, –OC(O)CH2CH2CH2CH2CH2).ATRP of DMAEMA from Br-PCL-Br
The difunctional macroinitiator Br-PCL-Br (40 mg, 8.0 μmol) was dissolved in toluene (0.20 mL). Into a Schlenk tube, charged with Cu(I)Br (3.8 mg, 26 μmol), the macroinitiator solution and DMAEMA (0.19 g, 1.2 mmol) were added followed by attachment of a septum. Two freeze–pump–thaw cycles were conducted, followed by the addition of HMTETA (23 mg, 0.10 mmol), and two more freeze–pump–thaw cycles were performed. The tube was immersed in an oil bath preheated to 70 °C, and the reaction was allowed to proceed for 1 h. Once the reaction had reached completion, the reaction mixture was allowed to cool down to ambient temperature under exposure to air, with subsequent addition of THF to dissolve the solid crude polymer. The polymer was precipitated into a 10-fold excess of cold heptane (−78 °C). The heptane was decanted; the polymer was dissolved in THF, transferred to a vial and concentrated under reduced pressure. It was then dried under reduced pressure at room temperature for 24 h. Yield: 0.20 g, 87%, calculations based on 100% conversion. The final product was denoted PDMAEMA-b-PCL-b-PDMAEMA, ABA, and characterised by NMR, SEC, DSC and DLS analysis. 1H NMR (400 MHz; CDCl3) δ (ppm): (PCL block) 4.00 (2H, m, –(CH2)4CH2OC(O)–), 2.25 (2H, m, –OC(O)CH2(CH2)4–), 1.59 (4H, m, –OC(O)CH2CH2CH2CH2CH2–), 1.32 (2H, m, –OC(O)CH2CH2CH2CH2CH2), (PDMAEMA blocks) 4.00 (2H, m, –CH2OC(O)C–), 2.51 (2H, m, –CH2N(CH3)2), 2.23 (6H, m, –(O)COCCH2CH2N(CH3)2), 1.86–1.77 (2H, s, –CH2C(C(O)CH3–)), 1.00–0.85 (3H, s, –CH2C(C(O)CH3–)).ATRP of DMAEMA
To a 50 mL round-bottomed flask, DMAEMA (20 g, 0.13 mol), acetone (20 g), HEBI (0.36 g, 1.7 mmol) and HMTETA (0.78 g, 3.4 mmol) were added. The flask was thereafter closed with a rubber septum and evacuated and back-filled with argon. Thereafter, the septum was carefully removed and Cu(I)Cl (0.17 mg, 1.7 mmol) was added, after which the septum was reattached and two more vacuum/argon cycles were performed. The flask was immersed in an oil bath preheated to 50 °C and the reaction was allowed to proceed for 45 minutes. After that time, the flask was removed from the oil bath, the septum was carefully removed, Cu(II)Br2 (0.10 mg, 0.45 mmol) was added, the septum was reattached, and the reaction mixture was left stirring at ambient temperature under argon flow for 20 minutes. The septum was removed and THF was added to dissolve the polymer formed. The polymer solution was passed through an activated, neutral Al2O3 column to remove the copper salt and was thereafter precipitated in a 10-fold excess of cold heptane (−78 °C). The heptane was decanted, the polymer dissolved in THF, transferred to a vial and concentrated under reduced pressure. It was then dried under reduced pressure at room temperature for 24 h. Yield: 7.4 g, 36%, calculations based on 100% conversion. The final product was denoted HO-PDMAEMA-Br, A-1, and characterised by NMR, SEC, FT-IR, DSC and MALDI-ToF analysis. 1H NMR (400 MHz; CDCl3) δ (ppm): 4.00 (2H, m, –CH2OC(O)C–), 2.51 (2H, m, –CH2N(CH3)2), 2.23 (6H, s, –(O)COCCH2CH2N(CH3)2), 1.90 (3H, s, –CH2C(C(O)CH3Br)), 1.86–1.77 (2H, s, –CH2C(C(O)CH3–)), 1.00–0.85 (3H, s, –CH2C(C(O)CH3–)).End-group conversion of HO-PDMAEMA-Br
A dried two-necked round-bottomed flask was charged with HO-PDMAEMA-Br (5.5 g, 1.6 mmol) and DMF (20 mL). After the polymer was dissolved, NaN3 (1.1 g, 16 mmol) was added and the flask was sealed with a rubber septum. The flask was immersed in an oil bath preheated to 80 °C and the reaction was left to proceed for 24 h. After the reaction was completed, the reaction mixture was allowed to cool down to ambient temperature. A 10-fold excess of deionised water was heated to 70 °C, and the reaction mixture was added drop-wise to the water and stirred for 10 min, upon which the product became insoluble and stuck to the walls. The water was decanted off, the product dissolved in THF, transferred to a vial and concentrated under reduced pressure. It was dried under reduced pressure at room temperature for 24 h. Yield: 3.1 g, 55%, calculations based on 100% conversion. The final product was denoted HO-PDMAEMA-N3, A-2, and characterised by NMR, FT-IR and SEC analysis. 1H NMR (400 MHz; CDCl3) δ (ppm): 4.00 (2H, m, –CH2OC(O)C–), 2.51 (2H, m, –CH2N(CH3)2), 2.23 (6H, s, –(O)COCCH2CH2N(CH3)2), 1.86–1.77 (2H, s, –CH2C(C(O)CH3–)), 1.38 (3H, s, –CH2C(C(O)CH3N3)), 1.00–0.85 (3H, s, –CH2C(C(O)CH3–)).End-group conversion of HO-PDMAEMA-N3
A Schlenk tube was charged with HO-PDMAEMA-N3 (1.0 g, 0.29 mmol), propargyl alcohol (22 mg, 0.37 mmol), Cu(I)Br (82 mg, 0.57 mmol) and DMF (12 mL), and sealed with a septum. All reactants were allowed to dissolve under stirring. Three freeze–pump–thaw cycles were performed. PMDETA (99 mg, 0.57 mmol) was degassed through three cycles of 5 minutes alternating evacuation and back-filling of argon, and it was then added to the tube. The tube was placed in an oil bath set to 25 °C, and the reaction was allowed to proceed for 24 h. In the next step, the septum was removed and THF was added. The polymer solution was passed through an activated, neutral Al2O3 column to remove the copper salt and was thereafter concentrated under reduced pressure at elevated temperature to remove the DMF. The polymer was dissolved in THF and precipitated twice in a 10-fold excess of cold heptane (−78 °C). The heptane was decanted, the polymer dissolved in THF, transferred to a vial and concentrated under reduced pressure. It was then dried under reduced pressure at room temperature for 24 h Yield: 0.49 g, 48%, calculations based on 100% conversion. The final product was denoted HO-PDMAEMA-OH, A-3, and characterised by NMR, FT-IR and SEC analyses. The product is 1H NMR (400 MHz; CDCl3) δ (ppm): 7.70 (1H, s, –CNCHC–), 4.71 (2H, m, HOCH2CCHNC–), 4.00 (2H, m, –CH2OC(O)C–), 2.51 (2H, m, –CH2N(CH3)2), 2.23 (6H, s, –(O)COCCH2CH2N(CH3)2), 1.86–1.77 (2H, s, –CH2C(C(O)CH3–)), 1.38 (3H, s, –CH2C(C(O)CH3N3C–)), 1.00–0.85 (3H, s, –CH2C(C(O)CH3–)).ROP of ε-CL from HO-PDMAEMA-OH
All glass equipment was dried prior to use in an oven at 150 °C. Into a two-necked round-bottomed flask Sn(Oct)2 (31 mg, 80 μmol) was introduced and dissolved in toluene (2.0 mL) under stirring for 30 min. HO-PDMAEMA-OH (0.30 g, 90 μmol) was dissolved in toluene (1.0 mL) in a glass vial. A Schlenk tube was charged with ε-CL (2.1 mL, 19 mmol), the solution of HO-PDMAEMA-OH and the solution of Sn(Oct)2, and the reaction mixture was stirred for 10 min at room temperature under a flow of argon. The tube was then immersed in an oil bath preheated to 62 °C. After 69 h, the reaction mixture was allowed to cool down to ambient temperature, exposed to air, dissolved in THF and precipitated in a 10-fold excess of cold MeOH (−20 °C). The polymer was recovered via filtration and then dried under reduced pressure at room temperature for 24 h. Yield: 1.0 g, 43%, calculations based on 100% conversion. The final product was denoted PCL-b-PDMAEMA-b-PCL, BAB, and characterised by NMR and SEC analysis. 1H NMR (400 MHz; CDCl3) δ (ppm): (PCL blocks) 4.00 (2H, m, –(CH2)4CH2OC(O)–), 2.25 (2H, m, –OC(O)CH2(CH2)4–), 1.59 (4H, m, –OC(O)CH2CH2CH2CH2CH2–), 1.32 (2H, m, –OC(O)CH2CH2CH2CH2CH2), (PDMAEMA block) 4.00 (2H, m, –CH2OC(O)C–), 2.51 (2H, m, –CH2N(CH3)2), 2.23 (6H, m, –(O)COCCH2CH2N(CH3)2), 1.86–1.77 (2H, s, –CH2C(C(O)CH3–)), 1.00–0.85 (3H, s, –CH2C(C(O)CH3–)).Purification attempts for the triblock PCL-b-PDMAEMA-b-PCL
The product was dissolved in THF and precipitation was attempted in a 10-fold excess of heptane (−78 °C). The product did not precipitate. The heptane was therefore concentrated and the polymer was dissolved in THF, followed by alternating ultrasonication for 5 minutes and precipitation of small amounts of polymer solution in a 10-fold excess of MeOH (−78 °C). The product was dissolved in DMF and precipitated in a 10-fold excess of ether (−78 °C). The product was dissolved in THF and fractionated precipitation was performed by slowly adding MeOH (RT) drop-wise. The product was subjected to separation attempts through Sephadex columns, both with THF and DMF as mobile phase followed by precipitation in a 10-fold excess of MeOH (−78 °C) and a 10-fold excess of ether (−78 °C), respectively. Finally, attempts to separate the product was performed through ultrafiltration (MWCO 10000 g mol−1) in DMF and precipitation in a 10-fold excess of ether (−78 °C). The fractions were characterised by NMR, SEC, DLS and DSC analysis.
Conversion of PDMAEMA into PMMA for SEC analysis
The procedure for converting PDMAEMA into poly(methyl methacrylate) (PMMA) was adopted from Plamper et al.52 and was divided into three parts: quaternisation, alkaline hydrolysis and alkylation.PDMAEMA (1.0 g, 6.4 mmol) was dissolved in THF (18 mL) in a 50 mL round-bottomed flask. MeI (1.1 g, 7.6 mmol) was dissolved in THF (5.0 mL), and added drop-wise to the PDMAEMA solution, the flask was sealed with a septum and the mixture left to stir overnight. The polymer solution was precipitated in a 10-fold excess of cold heptane (−78 °C), filtered and dried under reduced pressure at room temperature for 24 h. Yield: 0.40 g, 37%, calculations based on 100% conversion. The final product was denoted qPDMAEMA, and characterised by 1H-NMR analysis. 1H NMR (400 MHz; D2O) δ (ppm): 3.92 (2H, m, –(CH3)3NCH2CH2OC(O)C–), 3.35 (11H, m, –CH2N(CH3)3), 2.10 (2H, s, –CH2C(C(O)CH3–)), 1.21–1.09 (3H, s, –CH2C(C(O)CH3–)).
qPDMAEMA (0.40 g) was dissolved and stirred in a concentrated aqueous NaOH solution (8.0 mL) for 5 days at 90 °C. A dark precipitate was formed that was filtered off through a nylon filter. HCl was added drop-wise until the pH turned slightly acidic, determined with litmus paper, followed by freeze drying overnight to remove residual HCl. The product, poly(methacrylic acid) (PMAA), was dissolved in deionised water (8.0 mL) and subjected to aqueous dialysis (Millipore SpectraPore 7 MWCO 1000). To assure that the product was fully protonated, the steps from the addition of HCl to freeze drying were repeated. Yield: 85 mg, 21%, calculations based on 100% conversion. 1H-NMR analysis in D2O was conducted to assess the outcome of the hydrolysis. 1H NMR (400 MHz; D2O) δ (ppm): 2.10 (2H, s, –CH2C((C(O)OH)CH3–)), 1.21–1.09 (3H, s, –CH2C((C(O)OH)CH3)).
PMAA (30 mg) was dissolved in deionised water (0.40 mL) and THF (6.0 mL). Trimethyldiazomethane solution was added drop-wise while stirring until the yellow colour formed was stable for more than 1 hour. Subsequent dialysis in THF (Millipore SpectraPore 7 MWCO 1000) was performed, and drying of the resulting PMMA, C, at reduced pressure for 24 h. Yield: 10 mg, 24%, calculations based on 100% conversion. The final product was characterized by 1H-NMR and SEC analysis. 1H NMR (400 MHz; CDCl3) δ (ppm): 3.59 (3H, s, –CH2C(C(O)OCH3)CH3–), 1.86–1.77 (2H, s, –CH2C(C(O)OCH3)CH3–), 1.21–1.09 (3H, s, –CH2C(C(O)OCH3)CH3–).
Results and discussion
Scheme 1 depicts the synthesis of Br-PCL-Br, and the ABA-type triblock, PDMAEMA-b-PCL-b-PDMAEMA. In the first step, Sn(Oct)2-catalysed Ring-Opening Polymerisation (ROP) of ε-CL was initiated by the heterobifunctional HEBI, carrying a single Atom Transfer Radical Polymerisation (ATRP) initiating site and a hydroxyl-group. The polymerization resulted in a PCL-block with an ATRP-initiator at one chain-end and an OH-functionality at the other. In order to perform ATRP of DMAEMA from both ends of this polymer, a second ATRP initiating site was introduced by the reaction of the ω-hydroxyl function with BiB, creating Br-PCL-Br, B. The SEC trace of B exhibited a small shoulder on the high molecular weight side (Fig. 1), which was originating from the original precursor (Br-PCL-OH). Therefore, it is hypothesised that this most likely is attributed to transesterification reactions taking place during the ROP and not during the end-group transformation. 1H NMR experiment confirmed near to quantitative transformation of the hydroxyl-functional group into the ATRP initiator. | ||
| Scheme 1 Synthesis of the triblock PDMAEMA-b-PCL-b-PDMAEMA (ABA). Reagents and conditions: (i) ε-CL, Sn(Oct)2, THF, 62 °C; (ii) BiB, DMAP, TEA, THF, 25 °C; (iii) DMAEMA, Cu(I)Br, HMTETA, toluene, 70 °C. | ||
 | ||
| Fig. 1 SEC traces of Br-PCL-Br (B, solid line) and the triblock PDMAEMA-b-PCL-b-PDMAEMA (ABA, dashed line), acquired in DMF + 0.01 M LiBr. | ||
B was chain-extended with DMAEMA via Cu(I)Br/HMTETA-mediated ATRP in toluene at 70 °C (Scheme 1) to yield polymer ABA. Excess of HMTETA was employed to overcome the undesirable ligation of the copper salts with the monomer. Fairly high macroinitiator efficiency was perceived as the SEC trace underwent a clear shift to a lower elution volume (Fig. 1). Despite a slight tailoring of ABA, which has been observed for homopolymerisation and chain extension of PDMAEMA previously,51 ĐM of 1.24 confirmed good control over the reaction. The molecular weight characteristics of macroinitiators and block copolymers are summarised in Table 1.
![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif)
Successful formation of ABA was verified by 1H NMR experiment (Fig. 2, top). Resonances originating from both PCL and PDMAEMA blocks were observed. The notations a and s, added to f and g, designate whether the monomer sequence in the PDMAEMA backbone is atactic or syndiotactic, respectively.53 The degree of polymerization (DP) of the PDMAEMA blocks was estimated to be 58 by comparing the integrals of the peaks c and i, which were ascribed to PCL and PDMAEMA, respectively.
 | ||
| Fig. 2 1H-NMR spectra of the two triblocks PDMAEMA-b-PCL-b-PDMAEMA (ABA, top) and PCL-b-PDMAEMA-b-PCL (BAB, bottom) in CDCl3. Notations a and s added to f and g refer to tacticity; atactic and syndiotactic, respectively. | ||
Having obtained PDMAEMA-b-PCL-b-PDMAEMA triblock copolymer ABA (Scheme 1), the synthesis of PCL-b-PDMAEMA-b-PCL (BAB) was carried out according to Scheme 2. ATRP of DMAEMA was mediated by Cu(I)Cl/HMTETA catalytic complex, and chlorine atoms at the “living” termini were exchanged by bromine atoms by addition of Cu(II)Br2 at the end of the reaction. Thus, heterobifunctional HO-PDMAEMA-Br A-1 (Scheme 2) with the DP = 20 (estimated by NMR spectroscopy) and the ĐM = 1.25 was obtained in a controlled manner. The second hydroxyl-functional group was introduced via the Cu(I)-catalysed “click” reaction. Initially, nucleophilic substitution with NaN3 afforded HO-PDMAEMA-N3 A-2 (Scheme 2). Then A-2 was reacted with propargyl alcohol (1.3 eq.) yielding HO-PDMAEMA-OH, A-3 (Scheme 2). The end-group conversion from bromine to hydroxyl via azide was monitored and verified by NMR and FT-IR spectroscopy.
 | ||
| Scheme 2 Synthesis of the triblock PCL-b-PDMAEMA-b-PCL. Reagents and conditions: (i) DMAEMA, Cu(I)Cl, HMTETA, acetone, 50 °C; (ii) NaN3, DMF, 80 °C; (iii) propargyl alcohol, Cu(I)Br, PMDETA, DMF, 25 °C; (iv) ε-CL, Sn(Oct)2, toluene, 62 °C. | ||
The overlay of the 1H-NMR spectra of HO-PDMAEMA-Br A-1 (Scheme 2) and HO-PDMAEMA-N3 A-2 (Scheme 2) is depicted in Fig. 3. It is the subtle change – up field shift of the resonance signal assigned to the methyl group of the ultimate repeating unit (the shift of d′ to d′′) – that corroborates the success of the reaction.46 However, quantification of the extent of the reaction is made difficult by the overlapping resonance peaks. The 1H-NMR spectrum of the “click” reaction product, HO-PDMAEMA-OH A-3, is shown in Fig. 4. Methylene groups a and h – adjacent to the α- and ω-hydroxyl functions, respectively – can be identified. HSQC NMR experiment reveals that proton h at 4.68 ppm correlates with the carbon nucleus resonating at 55.86 ppm (Fig. S1†); thus it can be unambiguously attributed to the methylene group between the triazole ring and the hydroxyl function. Furthermore, in Fig. 5 FT-IR spectra of the end-group conversion HO-PDMAEMA-Br (A-1) to HO-PDMAEMA-OH (A-3) via HO-PDMAEMA-N3 (A-2) (Scheme 2) show the appearance and disappearance of the azide group (around 2100 cm−1)54 in the sample, supporting the conclusion of a successful conversion from A-1 to A-3. No other peaks in Fig. 5 undergo any changes. A-3 was chain extended via ROP of ε-CL catalysed by Sn(Oct)2 in toluene (Scheme 2, BAB). With 1H-NMR experiment (Fig. 2, bottom) resonance signals originating from both PCL and PDMAEMA blocks were observed and the DP of the PCL blocks was estimated to be 335 by comparing the integrals of peaks c and i, which were ascribed to PCL and PDMAEMA, respectively. This DP is corresponding to a total molecular weight of 41800, which is higher than the theoretical molecular weight, a difference that could be ascribed to the difference in solubility of PCL and PDMAEMA in CDCl3, hence PDMAEMA is shielded and less pronounced in NMR.
 | ||
| Fig. 3 1H-NMR spectra: conversion from HO-PDMAEMA-Br (top spectrum) to HO-PDMAEMA-N3 (bottom spectrum) in CDCl3. | ||
 | ||
| Fig. 4 1H-NMR spectrum of HO-PDMAEMA-OH (A-3) in CDCl3. | ||
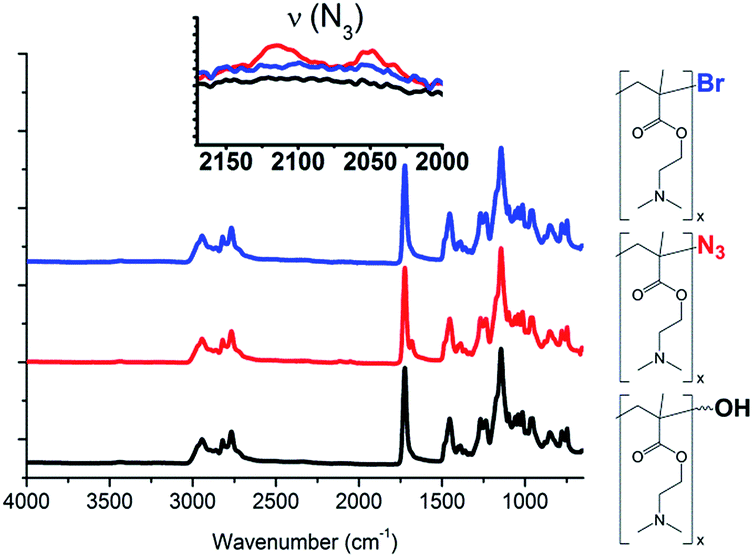 | ||
| Fig. 5 FT-IR spectra of products from end-group conversion experiments from bromide to azide and subsequent “click” reaction to yield OH-end group: HO-PDMAEMA-Br (A-1, blue), HO-PDMAEMA-N3 (A-2, red) and HO-PDMAEMA-OH (A-3, black). | ||
A clear shift of the elution volume in the SEC trace (Fig. 6) was detected, indicating a good macroinitiator efficiency, i.e., initiation occurred in both ends followed by efficient propagation of ε-CL. However, the trace was bimodal, giving the impression that the final product contained a mixture of di- and triblock copolymers. Therefore, several attempts were performed to purify the product and separate potential diblocks from the desired triblocks, utilising techniques such as: precipitation in different solvents, preparative SEC and ultrafiltration. However, all SEC traces of samples from the different purification techniques had the same appearance, i.e., the apparent bimodality remained. This is illustrated in Fig. 7 where all fractions from preparative SEC are displayed with an offset in intensity. The appearance of the SEC trace is independent of the separation technique, which indicates that the bimodality in the SEC traces is not caused by a mixture of di- and triblocks but an effect of a physical phenomenon occurring during SEC analysis.
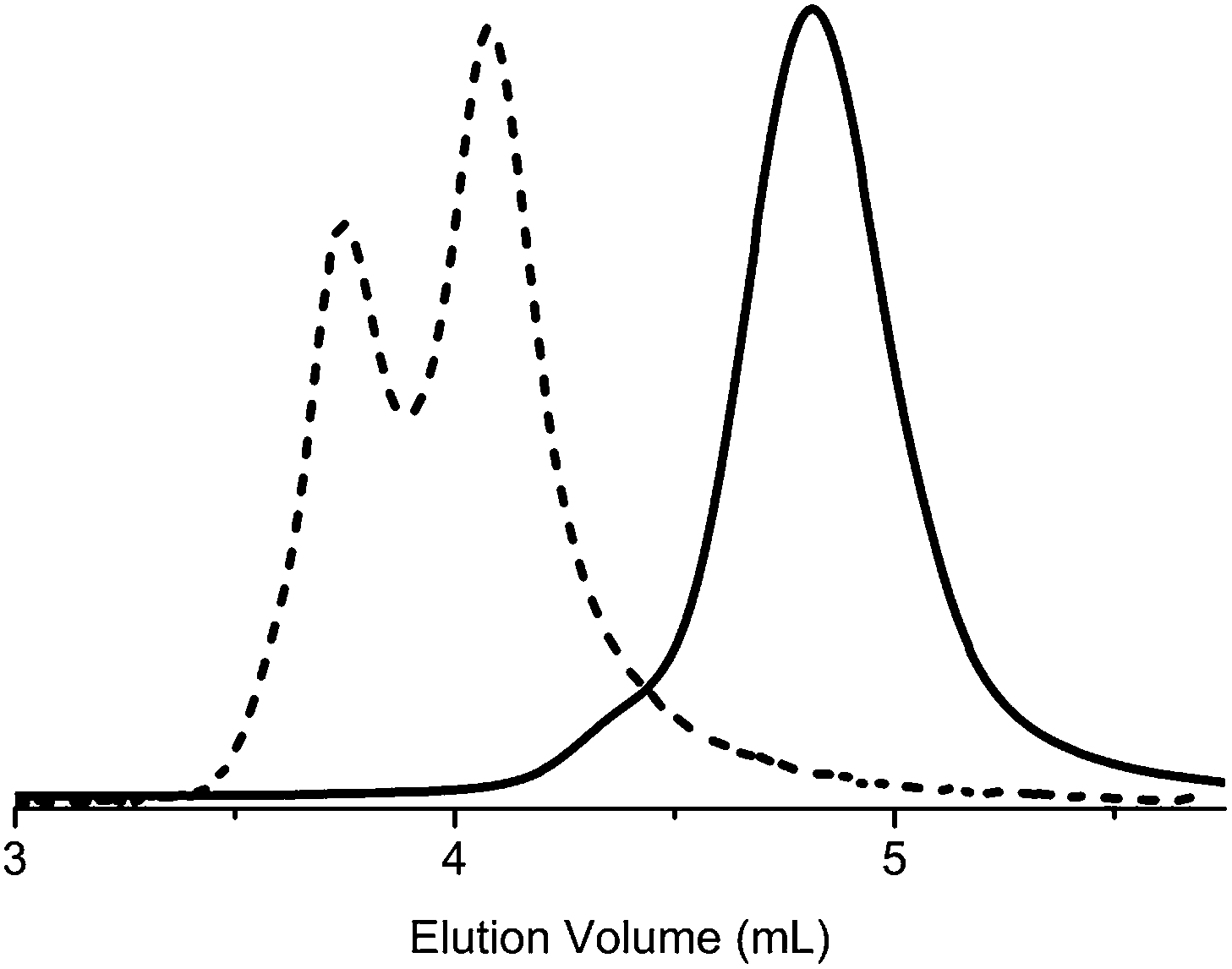 | ||
| Fig. 6 DMF-SEC traces of HO-PDMAEMA-OH (A-3, solid line) and triblock PCL-b-PDMAEMA-b-PCL (BAB, dashed line). | ||
 | ||
| Fig. 7 DMF-SEC traces of all fractions from preparative SEC of PCL-b-PDMAEMA-b-PCL (BAB). | ||
It has previously been stated that analysing PDMAEMA with THF SEC is difficult due to the potential adsorption of PDMAEMA onto the columns, giving rise to broadening of the peak (higher ĐM) and lower elution volume (higher molecular weight (Mw)).55 Additionally, PDMAEMA is a thermoresponsive polymer, exhibiting a lower critical solution temperature (LCST) in a temperature range of 38–80 °C in water depending on pH.56,57 Even though the mobile phase of the SEC system was DMF, this thermoresponsive behaviour could cause possible agglomeration since the SEC system was operated at 50 °C and DMF, as water, has the ability to form hydrogen bonds that can interact with PDMAEMA.58 To investigate the temperature dependence, DLS measurements were performed in DMF + 0.01 M LiBr, the solvent used for SEC, at 25 °C and 50 °C, as well as during heating in between those two temperatures, and it was found that the sizes of the spontaneously formed micelle-type structures varied for triblock BAB between 5–10 nm up to 5000 nm with increasing temperature (Fig. S3†).
Therefore, it is believed that the bimodal character of the trace in SEC (Fig. 6) for BAB is a phenomenon, not reflecting true values for ĐM and/or Mn, caused by plausible interaction with the SEC columns in combination with, and possibly enhanced by, temperature-induced agglomeration in the solvent used for the SEC analysis. Hence, it could be concluded that the targeted product, i.e. the triblock, had been obtained.
On the other hand, triblock ABA had a monomodal, slightly tailed, trace in SEC. Consequently, the fact that only a minor variation in size with temperature in DLS was observed was expected (Fig. S3†). It is, therefore, hypothesized that the temperature-induced effect in SEC and strong column interactions, yielding traces not reflecting true values for ĐM and/or Mw, is more pronounced with PDMAEMA being the middle block.
To further investigate the behaviour of PDMAEMA in SEC, the PDMAEMA was converted into PMMA (Scheme 3, C), through quaternisation, alkaline hydrolysis and subsequent methylation, allowing SEC analysis without plausible adsorption to columns or thermoresponsive behaviour. In Fig. 8 the DMF SEC (calibrated against PMMA) traces of PDMAEMA and the corresponding PMMA are displayed.
 | ||
| Scheme 3 Conversion of PDMAEMA into PMMA (C). Reagents and conditions: (i) MeI, THF, 25 °C; (ii) NaOH/H2O, HCl, 90 °C; (iii) trimethyldiazomethane solution, THF, H2O, 25 °C. | ||
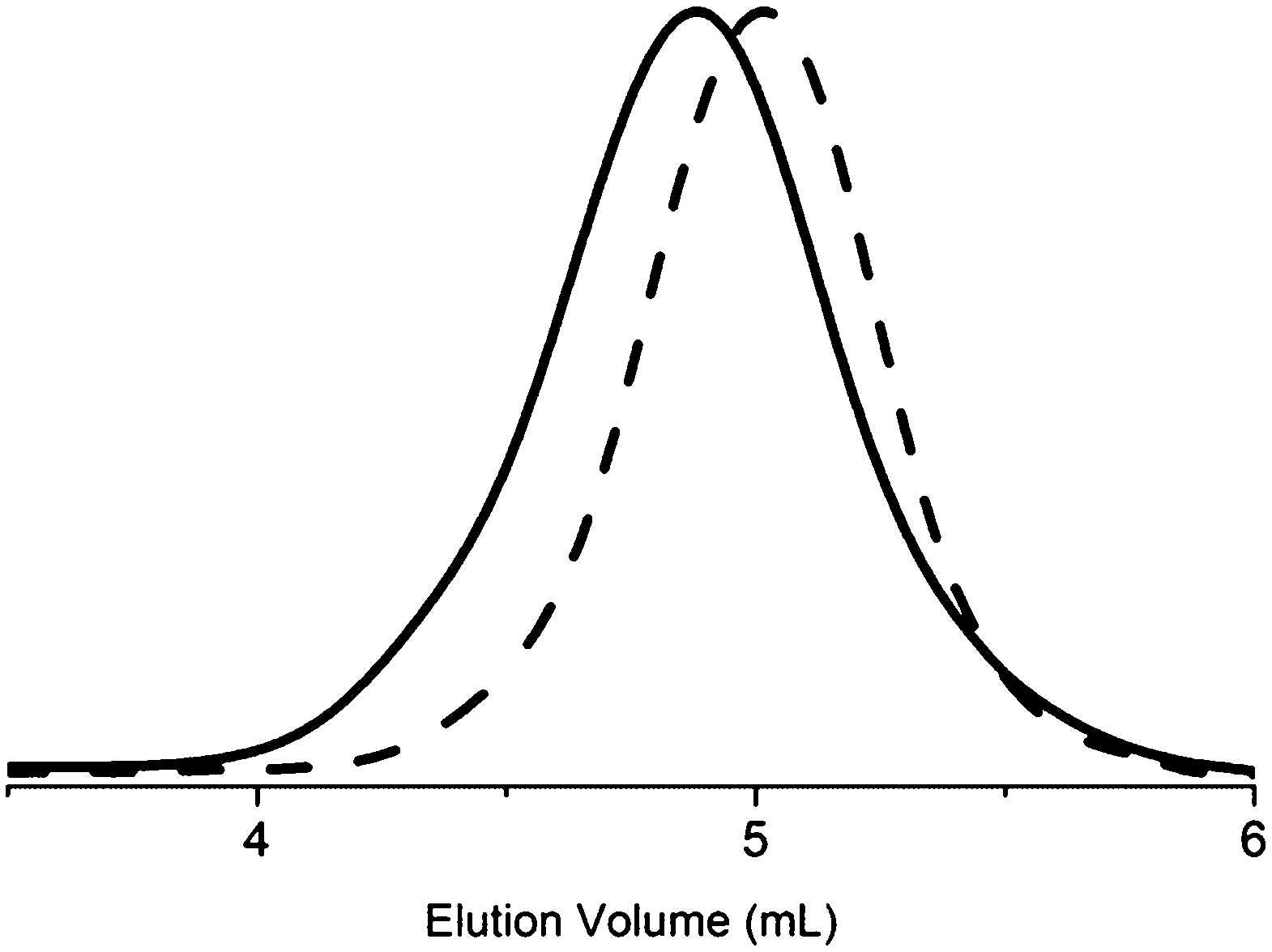 | ||
| Fig. 8 DMF-SEC traces of PDMAEMA before (solid line) and after conversion into PMMA (dashed line). | ||
The molecular weight was, as expected, somewhat lower, 5800 g mol−1 as compared with 7100 g mol−1 prior to conversion, since Mw of MMA is lower than that of DMAEMA and in theory the conversion should yield a PMMA with a Mw of 4500 g mol−1. The ĐM, on the other hand, was substantially lower, decreasing from 1.27 to 1.07, a difference ascribed to the influence of the dimethylamino group of PDMAEMA. This concludes that analysing PDMAEMA, or block copolymers of it, with DMF SEC at 50 °C gives a trace that to a greater extent compared with other linear polymers lacking this group, does not reflect the actual molecular weight and/or distribution.
PCL is a semicrystalline polymer with a glass transition temperature (Tg) of around −60 °C (ref. 59) and a melting temperature (Tm) of around 60 °C,60 while PDMAEMA is an amorphous polymer with a Tg of around 10 °C.61 The thermal properties of the macroinitiators and triblock copolymers were investigated with DSC, and the results are compiled in Table 2.
As expected, B exhibited a low Tg and a high crystallinity, close to the reported values for PCL. However, after formation of ABA the inherent crystallinity of PCL disappears while the Tg is increased, which is ascribed to the inherent characteristics of PDMAEMA. It is assumed that PDMAEMA prevents crystallisation, at the same time as it gives rise to a different packing of the polymer chains compared with the one that existed in B, yielding a higher Tg. From the Fox equation [eqn (2)], the weight fraction of PCL was estimated to be 36%, which is in line with 1H-NMR estimation since it would be equivalent to a total molecular weight of 22300 g mol−1. The thermal properties of A-3, no crystallinity and a Tg in the vicinity of 10 °C, are characteristic of the PDMAEMA homopolymer. In previous work, diblock copolymers of PCL and PDMAEMA were prepared and DSC analysis showed that they exhibited semi-crystallinity arising from the PCL block. Hence, the conclusion can therefore be drawn that it is the architecture of ABA triblock copolymer that obstructs crystallinity. On the other hand, in triblock BAB where PDMAEMA is the central block, a phase separation can be observed since there are two Tg's detected: one close to the Tg for B and one close to that for A-3. As a consequence, the Fox equation cannot be applied. Nevertheless, the inherent characteristics of PCL are pronounced in the form of exhibiting crystallinity and melting. Even though the crystallinity is slightly lower compared with PCL, B, it shows that the molecular architecture of these types of block copolymers is of high importance when tuning the degree of crystallinity/morphology.
Conclusions
Two types of triblocks of PDMAEMA and PCL have been prepared, ABA and BAB. PDMAEMA-b-PCL-b-PDMAEMA (ABA) was obtained through Sn(Oct)2-catalysed ROP of ε-CL followed by end-group transformation and Cu(I)Cl/HMTETA mediated ATRP of DMAEMA. PCL-b-PDMAEMA-b-PCL (BAB) was synthesised by ATRP of DMAEMA followed by end-group conversion through azide–alkyne “click” chemistry and subsequent ROP of ε-CL. All samples were analysed by size exclusion chromatography where it was found that the evaluation of PDMAEMA-containing polymers was difficult due to the thermoresponsivity of PDMAEMA, affecting the solubility of the polymer in the temperature range at which the SEC was operated. From thermal analysis it was determined that the triblocks mainly exhibited the character of the outer blocks; in PDMAEMA-b-PCL-b-PDMAEMA the inherent crystallinity of PCL was not present, and in PCL-b-PDMAEMA-b-PCL the crystallinity was approximately as high as for neat PCL.Acknowledgements
BiMaC Innovation and NordForsk Researcher Network are acknowledged for financial support.Notes and references
- J.-S. Wang and K. Matyjaszewski, Macromolecules, 1995, 28, 7901–7910 CrossRef CAS.
- H. Arslan, A. Pfaff, Y. Lu, P. Stepanek and A. H. Müller, Macromol. Biosci., 2013, 14, 81–91 CrossRef PubMed.
- A. M. Alhoranta, J. K. Lehtinen, A. O. Urtti, S. J. Butcher, V. O. Aseyev and H. J. Tenhu, Biomacromolecules, 2011, 12, 3213–3222 CrossRef CAS PubMed.
- D. M. Haddleton, C. B. Jasieczek, M. J. Hannon and A. J. Shooter, Macromolecules, 1997, 30, 2190–2193 CrossRef CAS.
- N. M. L. Hansen, D. M. Haddleton and S. Hvilsted, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 5770–5780 CrossRef CAS.
- A. de Cuendias, M. Le Hellaye, S. Lecommandoux, E. Cloutet and H. Cramail, J. Mater. Chem., 2005, 15, 3264–3267 RSC.
- K. Jankova, M. Bednarek and S. Hvilsted, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 3748–3759 CrossRef CAS.
- I. Javakhishvili, K. Jankova and S. Hvilsted, Polym. Chem., 2013, 4, 662–668 RSC.
- L. Valtola, A. Koponen, M. Karesoja, S. Hietala, A. Laukkanen, H. Tenhu and P. Denifl, Polymer, 2009, 50, 3103–3110 CrossRef CAS PubMed.
- K. Jankova and S. Hvilsted, Macromolecules, 2003, 36, 1753–1758 CrossRef CAS.
- A. Kajiwara, K. Matyjaszewski and M. Kamachi, Macromolecules, 1998, 31, 5695–5701 CrossRef CAS.
- K. Matyjaszewski, Y. Nakagawa and S. G. Gaynor, Macromol. Rapid Commun., 1997, 18, 1057–1066 CrossRef CAS.
- K. Yu, L. Zhang and A. Eisenberg, Langmuir, 1996, 12, 5980–5984 CrossRef CAS.
- S. E. Webber, J. Phys. Chem. B, 1998, 102, 2618–2626 CrossRef CAS.
- F. Checot, A. Brulet, J. Oberdisse, Y. Gnanou, O. Mondain-Monval and S. Lecommandoux, Langmuir, 2005, 21, 4308–4315 CrossRef CAS.
- R. Savic, A. Eisenberg and D. Maysinger, J. Drug Targeting, 2006, 14, 343–355 CrossRef CAS PubMed.
- C. Porsch, Y. Zhang, Å. Östlund, P. Damberg, C. Ducani, E. Malmström and A. M. Nyström, Part. Part. Syst. Charact., 2013, 30, 381–390 CrossRef CAS.
- M. R. Talingting, Y. Ma, C. Simmons and S. E. Webber, Langmuir, 2000, 16, 862–865 CrossRef CAS.
- J. Li, W.-D. He and X.-L. Sun, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 5156–5163 CrossRef CAS.
- G. S. Kwon and K. Kataoka, Adv. Drug Delivery Rev., 1995, 16, 295–309 CrossRef CAS.
- O. Colombani, M. Ruppel, F. Schubert, H. Zettl, D. V. Pergushov and A. H. E. Mueller, Macromolecules, 2007, 40, 4338–4350 CrossRef CAS.
- F. Kohori, K. Sakai, T. Aoyagi, M. Yokoyama, Y. Sakurai and T. Okano, J. Controlled Release, 1998, 55, 87–98 CrossRef CAS.
- W. Agut, A. Brulet, C. Schatz, D. Taton and S. Lecommandoux, Langmuir, 2010, 26, 10546–10554 CrossRef CAS PubMed.
- W. Agut, D. Taton and S. Lecommandoux, Macromolecules, 2007, 40, 5653–5661 CrossRef CAS.
- J. Gensel, E. Betthausen, C. Hasenöhrl, K. Trenkenschuh, M. Hund, F. Boulmedais, P. Schaaf, A. H. E. Müller and A. Fery, Soft Matter, 2011, 7, 11144–11153 RSC.
- A. P. Narrainen, S. Pascual and D. M. Haddleton, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 439–450 CrossRef CAS.
- F. L. Baines, N. C. Billingham and S. P. Armes, Macromolecules, 1996, 29, 3416–3420 CrossRef CAS.
- X. Zhang and K. Matyjaszewski, Macromolecules, 1999, 32, 1763–1766 CrossRef CAS.
- P. van de Wetering, N. J. Zuidam, M. J. van Steenbergen, O. A. G. J. van der Houwen, W. J. M. Underberg and W. E. Hennink, Macromolecules, 1998, 31, 8063–8068 CrossRef CAS.
- T. Endo, Y. Shibasaki and F. Sanda, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2190–2198 CrossRef CAS.
- I. Javakhishvili, W. H. Binder, S. Tanner and S. Hvilsted, Polym. Chem., 2010, 1, 506–513 RSC.
- I. Javakhishvili and S. Hvilsted, Polym. Chem., 2010, 1, 1650–1661 RSC.
- M. V. Walter, P. Lundberg, D. Hult, A. Hult and M. Malkoch, Polym. Chem., 2013, 4, 2680–2690 RSC.
- H. Lönnberg, L. Fogelström, Q. Zhou, A. Hult, L. Berglund and E. Malmström, Compos. Sci. Technol., 2011, 71, 9–12 CrossRef PubMed.
- H. Lönnberg, K. Larsson, T. Lindström, A. Hult and E. Malmström, ACS Appl. Mater. Interfaces, 2011, 3, 1426–1433 Search PubMed.
- N. Nordgren, L. Carlsson, H. Blomberg, A. Carlmark, E. Malmström and M. W. Rutland, Biomacromolecules, 2013, 14, 1003–1009 CrossRef CAS PubMed.
- F. Bougard, M. Jeusette, L. Mespouille, P. Dubois and R. Lazzaroni, Langmuir, 2007, 23, 2339–2345 CrossRef CAS PubMed.
- L. Mespouille, M. Vachaudez, F. Suriano, P. Gerbaux, O. Coulembier, P. Degée, R. Flammang and P. Dubois, Macromol. Rapid Commun., 2007, 28, 2151–2158 CrossRef CAS.
- S. Utsel, C. Bruce, T. Pettersson, L. Fogelström, A. Carlmark, E. Malmström and L. Wågberg, ACS Appl. Mater. Interfaces, 2012, 4, 6796–6807 CAS.
- S. Motala-Timol and D. Jhurry, Eur. Polym. J., 2007, 43, 3042–3049 CrossRef CAS PubMed.
- C. Zhu, S. Jung, S. Luo, F. Meng, X. Zhu, T. G. Park and Z. Zhong, Biomaterials, 2010, 31, 2408–2416 CrossRef CAS PubMed.
- G. Siqueira, J. Bras and A. Dufresne, Polymer, 2010, 2, 728–765 CAS.
- D. Klemm, F. Kramer, S. Moritz, T. Lindstrom, M. Ankerfors, D. Gray and A. Dorris, Angew. Chem., Int. Ed. Engl., 2011, 50, 5438–5466 CrossRef CAS PubMed.
- A. K. Bledzki and J. Gassan, Prog. Polym. Sci., 1999, 24, 221–274 CrossRef CAS.
- J. George, M. S. Sreekala and S. Thomas, Polym. Eng. Sci., 2001, 41, 1471–1485 CAS.
- V. Coessens and K. Matyjaszewski, J. Macromol. Sci., Part A: Pure Appl. Chem., 1999, 36, 667–679 CrossRef PubMed.
- R. Huisgen, G. Szeimies and L. Moebius, Chem. Ber., 1967, 100, 2494–2507 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- B. Wunderlich, Macromolecular Physics, Academic Press, New York, 1980, vol. 3, Crystal Melting Search PubMed.
- T. G. Fox, Bull. Am. Phys. Soc., 1956, 1, 123 CAS.
- R. A. Cordeiro, N. Rocha, J. P. Mendes, K. Matyjaszewski, T. Guliashvili, A. C. Serra and J. F. J. Coelho, Polym. Chem., 2013, 4, 3088–3097 RSC.
- F. A. Plamper, A. Schmalz, E. Penott-Chang, M. Drechsler, A. Jusufi, M. Ballauff and A. H. E. Müller, Macromolecules, 2007, 40, 5689–5697 CrossRef CAS.
- J. Huang, R. R. Koepsel, H. Murata, W. Wu, S. B. Lee, T. Kowalewski, A. J. Russell and K. Matyjaszewski, Langmuir, 2008, 24, 6785–6795 CrossRef CAS PubMed.
- N. V. Tsarevsky, S. A. Bencherif and K. Matyjaszewski, Macromolecules, 2007, 40, 4439–4445 CrossRef CAS.
- A. P. Narrainen, S. Pascual and D. M. Haddleton, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 439–450 CrossRef CAS.
- J. Niskanen, C. Wu, M. Ostrowski, G. G. Fuller, S. Hietala and H. Tenhu, Macromolecules, 2013, 46, 2331–2340 CrossRef CAS.
- F. A. Plamper, M. Ruppel, A. Schmalz, O. Borisov, M. Ballauff and A. H. E. Müller, Macromolecules, 2007, 40, 8361–8366 CrossRef CAS.
- Y. J. Chang and E. W. Castner, J. Chem. Phys., 1993, 99, 113–125 CrossRef CAS PubMed.
- C. Allen, D. Maysinger and A. Eisenberg, Colloids Surf., B, 1999, 16, 3–27 CrossRef CAS.
- E. Núñez, C. Ferrando, E. Malmström, H. Claesson, P. E. Werner and U. W. Gedde, Polymer, 2004, 45, 5251–5263 CrossRef PubMed.
- R. L. Teoh, K. B. Guice and Y.-L. Loo, Macromolecules, 2006, 39, 8609–8615 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: HSQC NMR spectrum of HO-PDMAEMA-OH, MALDI-TOF spectra of HO-PDMAEMA-OH and Br-PCL-Br, and DLS measurements of triblock copolymers. See DOI: 10.1039/c4ra04325a |
| This journal is © The Royal Society of Chemistry 2014 |