Cu(II) PBS-bridged PMOs catalyzed one-pot synthesis of 1,4-disubstituted 1,2,3-triazoles in water through click chemistry†
Avvari N. Prasadab,
Benjaram M. Reddy*a,
Eun-Young Jeongb and
Sang-Eon Park*b
aInorganic and Physical Chemistry Division, CSIR–Indian Institute of Chemical Technology, Hyderabad – 500007, India. E-mail: bmreddy@iict.res.in; Tel: +91 40 27193510
bLaboratory of Nano-Green Catalysis, Department of Chemistry, Inha University, 253 Yonghyun-dong, Incheon 402-751, Republic of Korea. E-mail: separk@inha.ac.kr; Tel: +82 32 860 7675
First published on 11th June 2014
Abstract
A series of PBS-HPMO and Cu(II)-PBS-HPMO were synthesized from the self-assembly of 1,2-bis(triethoxysilyl)ethane and porphyrin-bridged silsesquioxane (PBS). These synthesized PBS-HPMO and Cu(II)-PBS-HPMO were characterized using different spectroscopic and non-spectroscopic techniques, namely, XRD, FT-IR spectroscopy, nitrogen adsorption–desorption isotherms, and UV-visible and EPR spectroscopies. Among these, the porphyrin-bridged PMOs, specifically Cu(II)-PBS-HPMO, were found to be proficient catalysts for the multicomponent reaction of benzyl halides with sodium azide and terminal alkynes. This catalyst allowed for the high regioselective synthesis of 1,4-disubstituted 1,2,3-triazoles through a one-step and atom economic tandem reaction with water as the solvent. Note that no additional base or ligand or reducing agent is required. Moreover, in addition to benzyl halides, hetero benzyl halides have also been achieved in remarkable yields and in a completely regioselective manner. A series of structurally diverse 1,2,3-triazoles were also prepared in good to excellent yields from easily accessible starting materials by employing this protocol. Furthermore, this process is purely heterogeneous and the cascade reactions were performed in water, and the efficient catalyst recyclability makes such a synthesis a truly green process.
1 Introduction
In the context of green chemistry, the search for alternative safer, cleaner, and ecofriendly technologies plays a vital role in synthetic organic chemistry. With this objective, chemists are facing major challenges to develop organic chemistry processes using new catalysts and catalytic systems that result in less waste and generate fewer hazardous substances. Catalysis, in particular, heterogeneous catalysis is one of the fundamental aspects of green chemistry. Multicomponent reactions (MCRs) have proven to be very prominent and effective bond-forming tools in synthetic organic chemistry along with medicinal and combinatorial chemistry, in recent years. MCRs are convergent chemical synthesis processes, in which three or more starting materials are involved in a single synthetic operation without any isolation of any intermediate. These approaches have multiple advantages such as the elimination of complicated purification operations, use of readily available starting materials and permit savings on solvents as well as reagents. In addition, MCRs are the most potent tools for the creation of intricate structures in a single step.1,2 Hence, we have been working on MCRs, employing a new class of ecofriendly heterogeneous catalysts, which are easy to prepare, handle, and recycle.Since 2001, the click reaction of azides and terminal alkynes, which was pioneered by Sharpless and co-investigators, has received prominent attention.3 Huisgen 1,3-dipolar azide–alkyne cycloaddition4 has been recognized as one of the most noteworthy synthetic tools because of the expedient and reliable assembly of 1,2,3-triazole moieties, which exhibit a wide spectra of biological activities and are thus often found to be the vital nuclei of biologically active molecules.5 Moreover, the cycloaddition of azides with alkynes has emerged as an archetypical example of “click chemistry,” and it is a key step in the first series of click backbone amide linkers.6 The obtained 1,2,3-triazole scaffolds possess interesting properties and have led to a substantial growth in click chemistry research. The wide scope of azide–alkyne cycloaddition is demonstrated by its use in numerous areas of life and material sciences, including drug discovery,7 bioconjugation chemistry,8 supramolecular chemistry,9 solid phase reactions,10 DNA labelling,11 drug-like molecules with significant biological properties comprising anti-HIV activity,12 as well as antimicrobial activity against Gram-positive bacteria, the assembly of glycoclusters13 and glycodendrimers,14 and for stationary phases in functionalized HPLC columns.15
In heterocyclic chemistry, the triazole moiety is treated as a robust keystone in complex molecular architectures.16 Recently, several groups have reported triazole moieties acting as donors in metal complexes, including in applications in catalysis17 such as the tris-(triazole) ligands,18 and as very proficient catalysts for the azide–alkyne cycloaddition reaction. Further, triazolylmethyl phosphines and tetradentate bis-(triazolylamines) have been used for Pd-catalyzed allylic alkylation reactions19 and Mn-catalyzed epoxidation reactions,20 respectively. In addition, 1,2,3-triazole scaffolds are smart linking units due to their capability of hydrogen bonding; moreover, they are stable to metabolic degradation, which is responsible for binding the biomolecular targets and can improve the solubility of the compounds. The triazole moieties also display a number of chemotherapeutic properties along with significant anticonvulsant properties.21
The Cu(I) catalyzed cycloaddition of azides with alkynes has gained significant attention since its discovery by Sharpless and co-workers,3,22 as well as independently by Meldal et al.,23 in which copper(I) catalysts showed a dramatic rate acceleration of up to 107 times.24 Generally, the sources of copper(I) include the oxidation of copper metal turnings,25 the direct addition of cuprous salts (CuI, CuBr) with methyl(phenyl)sulfane,26 iminopyridine,27 mono- (or) polydentate nitrogen ligands,18,28 N-heterocyclic copper carbene complexes,22b and the comproportionation of copper(0)/copper(II), which is usually limited to special applications (e.g., biological systems).29 Recently, copper catalysts immobilized on various supports have been explored for the Huisgen cyclization of azides to alkynes, namely, amine-bound silica,30 superparamagnetic mesoporous silica,31 a ligand-bound organic polymer,32 activated charcoal,33 polysaccharide,34 zeolites,35 hydrotalcite,36 and AlO(OH).37 Conversely, most of these catalysts with Cu(I) species often less advised due to their thermodynamic instability, and due to the formation of an alkyne–alkyne coupling product and other undesired by-products. In addition, many of these reports involved reactions that were homogeneous in nature, and required an inert atmosphere as well as anhydrous solvents.36a
Periodic mesoporous organosilicas (PMOs) have gained significant attention due to their large surface area and structural diversity of the organosilica frameworks. PMOs are an innovative type of organic–inorganic hybrid materials, synthesized from up to 100% organic-bridged alkoxysilane precursors (R–[Si(OR′)3]n, n ≥ 2, R = organic group, R′ = –CH3, –C2H5.), in which these organic groups are densely and covalently embedded within the silica framework.38 Moreover, in PMOs the organic groups are not only distributed uniformly inside the framework but also do not affect the pore, which is more favorable for guest molecule diffusion.39 PMOs with dissimilar bridging organic groups could be potentially applied in various fields, namely, adsorption, catalysis, drug delivery, metal ion detection, and optics.40 Specifically, PMOs are well suited for use as catalysts because of the numerous advantages they offer, including easy accessibility, rapid diffusion, and favorable mass transfer for substrates into and out of the mesopores.41 Hence, PMOs are promising candidates for heterogeneous catalysis, and they also offer good reusability owing to their strong covalent attachment of the catalytic active metal centres at the mesopore surface.42
We herein report the synthesis of a series of PBS-HPMO and Cu(II)-PBS-HPMO as novel catalysts, and their use for the synthesis of 1,4-disubstituted 1,2,3-triazoles as MCRs with readily available starting materials and water as the solvent. To the best of our knowledge, in only very few instances, has Cu(II) (as Cu(II)-hydrotalcite, Cu(II) acetate) been used for the regioselective synthesis of 1,4-disubstituted 1,2,3-triazoles.36 It is worth mentioning here that the Cu(II)-PBS-HPMO has been employed for the first time in the field of azide–alkyne cycloaddition MCR and has achieved a high product yield.
2 Experimental section
2.1 Reagents and materials
All chemicals for the synthesis of materials and catalytic activity testing were purchased from Sigma-Aldrich Corporation, St. Louis, MO (U.S.A.) and directly used without further purification.2.2 Synthesis of tetrakis(4-carboxyphenyl)porphyrin (TCPP)43
Pyrrole (10 mmol) and 4-carboxybenzaldehyde (10 mmol) were added to reagent grade propionic acid (100 mL). The mixture was refluxed for 1 h, and the resultant crude product was allowed to cool to room temperature, and then methanol (100 mL) was added and the solution was further chilled in an ice bath with stirring. Then, the resultant solid was removed by filtration and thoroughly washed with methanol and hot water, and then the porphyrin was purified. The observed atomic ratio of C/N was 12.35. The theoretical atomic ratio of C/N for {C48H30N4O8} is 12. The spectral data are as follows: 1H NMR (d6-DMSO): 8.85 ppm (s, 8H), 8.39–8.33 ppm (m, 16H). m/z 791 ([M + H]+ calcd for 790.77). FT-IR spectrum: 1299 cm−1 (C–O stretch of carboxylic acid), 1693 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O in carboxylic acid), 2500 cm−1 ∼3300 cm−1 (broad, (–OH) in acid).
O in carboxylic acid), 2500 cm−1 ∼3300 cm−1 (broad, (–OH) in acid).
2.3 Synthesis of porphyrin bridged silsesquioxane (PBS)
A mixture of TCPP (1 mmol), 3-aminopropyltriethoxysilane (4 mmol) and dicyclohexyl carbodiimide (4 mmol), THF (50 mL) were stirred at 80 °C for 12 h under a nitrogen atmosphere. The mixture was allowed to cool to room temperature. The reaction mixture was filtered and washed with THF and petroleum ether several times.43b The observed C/N was 9.82, and the theoretical atomic ratio of C/N for {C84H114N8O16Si4} is 10.5. The spectral data are as follows: 13C CPTOSS NMR: 10.46, 22.0, 31.12 and 42.02 ppm (C in propyl groups of silane and terminal CH3 of ethoxy group), 116, 127,134 and 143 ppm (aromatic and pyrrole C) and 173 ppm (CO in amide) FT-IR spectrum: 950–1250 cm−1 (Si–O–C), 1664 cm−1 (CO in amide), 1528, 3276 cm−1 (N–H bond).
2.4 PBS bridged hybrid periodic mesoporous organosilica (PBS-HPMO)
A series of PBS-HPMO were prepared by considering various molar percentages of PBS precursors. For this procedure, hexadecyltrimethylammonium bromide (CTAB, 0.47 g) was added to distilled water (8.85 g)–NH3H2O (4.45 g), and the mixture was vigorously stirred to obtain a clear solution (Table 1). Then, PBS and 1,2-bis(triethoxysilyl)ethane (BTEE) were added to the CTAB solution according to the desired ratio and the mixture was stirred for 24 h at room temperature. The resultant precipitate was collected by filtration, extracted with ethanol–HCl solution and dried at 60 °C.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Parameters for hybrid mesoporous organosilica synthesis using PBS and BTEE
Parameters for hybrid mesoporous organosilica synthesis using PBS and BTEE
| Catalyst | CTAB (g) | PBS (g) | BTEE (g) | Ammonia water–H2O |
|---|---|---|---|---|
| PBS-HPMO-1 | 0.47 | 0.02 | 0.9 | 4.45/8.85 |
| PBS-HPMO-2 | 0.47 | 0.08 | 0.9 | 4.45/8.85 |
2.5 Metalation of PBS-HPMO with copper
PBS-HPMOs (0.5 g) and copper acetate monohydrate (0.25 g) were added to methanol (30 mL), and the mixture was stirred for 12 h at 80 °C. The resultant precipitate was collected by filtration, washed with methanol and water several times, and dried at 60 °C (Scheme 1). The synthesized Cu-PBS-HPMOs were characterized by UV-Vis spectroscopy and electron paramagnetic resonance (EPR) spectroscopy. | ||
| Scheme 1 Typical synthetic route of Cu(II)-PBS-HPMOs. | ||
2.6 Characterization techniques
The X-ray diffraction (XRD) patterns were recorded on a Rigaku Multiflex diffractometer with a monochromated high-intensity Cu Kα radiation (λ = 1.54 Å). All samples were scanned under the ambient conditions over the 2θ range of 0.7–5° at a rate of 0.5° min−1 (40 kV, 30 mA). Solid-state NMR spectra were collected through a DSX Bruker NMR 600 MHz. The N2 adsorption–desorption isotherms and pore-size distribution were achieved by using a Micromeritics tristar apparatus at liquid N2 temperature. The specific surface area is obtained from a nitrogen adsorption isotherm by using the BET equation. The pore diameter was calculated by using the Barrett–Joyner–Halenda (BJH) method using the desorption branch of the adsorption–desorption isotherms. Cu(II)-PBS-PMOs were analysed and characterized by UV-visible and EPR spectroscopy by using Solidspec-3700 and JEOL FA200 instruments, respectively. FT-IR spectra were obtained by a VERTEX 80V FT-IR vacuum spectrometer using KBr pellets.2.7 Synthesis of 1,4-disubstituted 1,2,3-triazoles
All chemicals employed in this study were commercially available and used without further purification. A mixture of benzyl halide (1 mmol), sodium azide (1.2 mmol), terminal alkyne (1.1 mmol) and a catalytic amount of Cu(II)-PBS-HPMO-2 (10 mg) in water (1.5 mL) were stirred at 100 °C for an appropriate time. After completion of the reaction, as confirmed by thin layer chromatography (TLC), the reaction mixture was filtered off and washed with ethyl acetate (3 × 5 mL). The combined layers were dried over anhydrous Na2SO4, concentrated in vacuum and purified by column chromatography on 60–120 mesh silica gel using ethyl acetate and hexane as the eluent (1:9) to afford the pure 1,4-disubstituted 1,2,3-triazoles. All products were identified by comparing their spectral data with the literature. Moreover, the solid catalyst was conveniently separated by centrifugation from the reaction mixture, and its activity was examined in the subsequent experiments.
3 Results and discussion
The low-angle powder XRD patterns of the synthesized PBS-HPMO-n and Cu(II)-PBS-HPMO-n catalysts are presented in Fig. 1. As shown in Fig. 1, the well-resolved diffraction peaks at 2θ = 1.4–2° reflect the (100) planes of the ordered and textural uniformity of the MCM-41 type mesoporous materials. The N2 adsorption–desorption isotherms of PBS-HPMO-n are shown in Fig. 2. The isotherms can be classified as type IV isotherms, which correspond to the presence of ordered mesoporous materials.41,43 The textural properties, including the surface area, and pore diameter, are shown in Table 2. As the PBS content increases, the pore diameter curve shifts from 2.25 to 2.40 nm. The tendency of mesopore shrinkage with increasing PBS loading is also revealed by the changes in the surface area and total pore volume. | ||
| Fig. 1 Powder XRD patterns of PBS-HPMO-1 (a), PBS-HPMO-2 and Cu-PBS-HPMO-2 (b). | ||
 | ||
| Fig. 2 Nitrogen adsorption–desorption isotherms (a) and pore-size distribution (b) of PBS HPMO-n. | ||
From UV-vis spectroscopy, the spectra of the PBS-HPMO-1 and PBS-HPMO-2 show one Soret band at 419 and 418.5 nm, along with four Q bands at 516, 555, 597, 660 and 517, 553, 594, 647 nm. Moreover, Cu(II)-PBS-HPMO-1 and Cu(II)-PBS-HPMO-2 exhibit Soret bands at 410 and 409 nm and are accompanied with Q bands at 541 and 581 nm, respectively (Fig. 3). These obtained results revealed that the synthesized catalysts have the PBS unit in the bridged mesoporous organosilica (Table 3). Moreover, the change in the Q band region (fewer bands) on metalation is because of the changed symmetry of the free-base porphyrin.44
 | ||
| Fig. 3 UV-vis spectra of PBS (a) and PBS-PMO-n (b), and Cu-PBS-PMO-n (c). | ||
| Catalyst | Soret band (nm) | Q bands (nm) |
|---|---|---|
| PBS-HPMO-1 | 419 | 516, 555, 597, 660 |
| PBS-HPMO-2 | 418.5 | 517, 553, 594, 647 |
| Cu PBS-HPMO-1 | 410 | 541 |
| CuPBS-HPMO-2 | 409 | 541, 581 |
The typical FT-IR spectra of synthesized PBS-HPMO-n and Cu(II)-PBS-HPMO-n catalysts are presented in Fig. 4. The FT-IR spectra display a strong absorbance in the range of 1200–1000 cm−1 that reveal the stretching of the Si–O–Si bond, and the bands related to Si–O–C and Si–C overlap with the strong absorption bands of the Si–O–Si vibration modes in this region.45 All samples also show bands at 1644 cm−1 and 3443 cm−1, which are caused by the PBS moiety.
 | ||
| Fig. 4 FT-IR spectra of PBS-PMO-n (a) and Cu-PBS-PMO-n (b). | ||
Synthesized PBS-HPMOs and Cu(II)-PBS-HPMOs were characterized by means of EPR spectroscopy to confirm the presence of Cu(II) ions inside the PBS-HPMOs after metalation (Fig. 5). From Fig. 5, the EPR spectrum reveals the presence of Cu(II) ions in the synthesized materials, and the Cu(II)-PBS-HPMO-2 sample exhibits a higher intensity of signal than the Cu(II)-PBS-HPMO-1 due to smaller amount of Cu(II) ions inside the porphyrin-bridged PMO. The EPR spectrum of Cu(II)-PBS-HPMO-2 containing adsorbed Cu(II) ions shows the g parameters at g‖ ∼ 2.20 and g⊥ ∼ 2.06, which indicates that the Cu(II) ions are linked to nitrogen ligands in the PBS. Solid-state 29Si NMR experiments often afford expedient information regarding the chemical environment around the Si nuclei with the presence of organic functional moieties in the hybrid matrices. From the 29Si NMR spectra, the downfield chemical shifts at −64 and −57 ppm resemble different Si states (T2 and T3) such as T2 [SiR(OH)(OSi)2] and T3 [SiR(OSi)3] and are shown in Fig. 6, where R is the PBS moiety and BTEE, respectively. All characterization techniques revealed the formation of PBS-HPMOs and Cu(II)-PBS-HPMOs materials.
 | ||
| Fig. 5 EPR spectra of PBS-PMO-n (a) and Cu-PBS-PMO-n (b). | ||
 | ||
| Fig. 6 Solid-state 29Si NMR spectra of PBS-HPMO-2. | ||
As part of our ongoing research, we have been working on porphyrin-bridged PMOs having a regular porous structure, large surface area and tunable surface properties, as these play vital roles in the field of catalysis and are preferable for specific and selective catalysis. Very recently, we have successfully employed PMO catalysts for the Baeyer–Villiger oxidation reaction with cyclic ketones as the starting materials.41 We wish to extend our work to MCRs by using these PMOs; thus, we report herein a novel Cu(II)-PBS-bridged PMOs for MCRs using readily available starting materials such as benzyl halides, sodium azide, and terminal alkynes for the synthesis of 1,4-disubstituted 1,2,3-triazoles (Scheme 2). The MCR consists of a one-pot, two-step synthesis of 1,4-disubstituted 1,2,3-triazoles proceeded by a click reaction through the formation of benzyl azides from sodium azide and benzyl halides. In click chemistry, the formation of organic azide [R-N3] is the crucial step, and these azides made a fleeting appearance in organic synthesis. The benzyl azides are formed easily within a short reaction time and are confirmed by the observation of the characteristic azido stretching frequency at ∼2100 cm−1.36b Remarkably, the construction of heterocyclic frameworks with a nitrogen atom plays a vital role in organic synthesis, as well as in medicinal chemistry, because of their existence in many natural products and biologically active molecules.46
 | ||
| Scheme 2 Synthesis of 1,4-disubstituted 1,2,3-triazoles with different benzyl halides, sodium azide and various terminal alkynes by using Cu(II)-PBS-HPMO-2 catalyst. | ||
In order to determine the best catalytic system suited for the synthesis of 1,4-disubstituted 1,2,3-triazoles, we examined typical reaction parameters. For instance, benzyl bromide (1 mmol), sodium azide (1.2 mmol), and phenylacetylene (1.1 mmol) were used as model substrates with a series of synthesized PBS-HPMO, Cu(II)-PBS-HPMO catalysts in various solvents. These results are illustrated in Table 4. The preliminary results revealed that the Cu(II)-PBS-HPMO-2 catalyst exhibits promising catalytic activity in the presence of water under reflux conditions with enhanced regioselectivity towards the desired 1,4-disubstituted 1,2,3-triazole product (Table 4, Entry 4). The Cu(II)-PBS-HPMO-1 also showed good catalytic activity in comparison to copper-free PBS-HPMO catalyst (Table 4, Entry 3). Nevertheless, the yields were lower than that of the Cu(II)-PBS-HPMO-2 catalyst. The CuII species present in the frameworks of PMOs exhibited remarkable activity in the absence of an inert atmosphere and without the addition of any sacrificial ligands and additives. Trace amounts of products, including some by-products, and longer reaction times were noted in the absence of the catalyst (Table 4, Entry 5), and we also observed different spots in the TLC in the presence of catalyst under room temperature conditions (Table 4, Entry 7).
| Entry | Catalyst | Solvent | Temp. (°C) | Time (h) | Yieldb (%) |
|---|---|---|---|---|---|
| a Reagents and reaction conditions: benzyl bromide (1 mmol), sodium azide (1.2 mmol), phenylacetylene (1.1 mmol), catalyst (10 mg), and solvent (1.5 mL); unless mentioned otherwise.b Yields of isolated products.c Without catalyst.d Undesired products, including starting materials. | |||||
| 1 | PBS-HPMO-1 | Water | 100 | 7 | 45 |
| 2 | PBS-HPMO-2 | Water | 100 | 7 | 60 |
| 3 | Cu-PBS-HPMO-1 | Water | 100 | 4 | 81 |
| 4 | Cu-PBS-HPMO-2 | Water | 100 | 3.5 | 96 |
| 5 | — | Water | 100 | 10 | —c |
| 6 | Cu-PBS-HPMO-2 | — | 100 | 6 | 72 |
| 7 | Cu-PBS-HPMO-2 | Water | RT | 10 | —d |
| 8 | Cu-PBS-HPMO-2 | Toluene | 110 | 3.5 | 27 |
| 9 | Cu-PBS-HPMO-2 | Methanol | 65 | 3.5 | 70 |
| 10 | Cu-PBS-HPMO-2 | DCM | 40 | 3.5 | 15 |
| 11 | Cu-PBS-HPMO-2 | DMF | 120 | 3.5 | 65 |
| 12 | Cu-PBS-HPMO-2 | Benzene | 80 | 3.5 | 32 |
| 13 | Cu-PBS-HPMO-2 | THF | 65 | 3.5 | 64 |
| 14 | Cu-PBS-HPMO-2 | Acetonitrile | 80 | 3.5 | 73 |
After selection of the most efficient catalyst, we further screened with different solvents to adjust the reaction conditions. Among all the solvents, the reaction proceeded well in water, which was found to be the most appropriate solvent for the synthesis of desired 1,4-disubstituted 1,2,3-triazoles with excellent yields. The enriched product yields are partly due to the hydrophobic nature of the organic reactants, since their repulsion from water enhances the number of collisions between the organic molecules and increases their ground-state energies, leading to an increase in the reaction rate.47 In recent years, water has been considered as an excellent reaction medium in synthetic organic chemistry with many promising advantages. Moreover, we have observed that several reactions frequently progress optimally in pure water. Although, the rate acceleration is trivial, the use of water as the only reaction medium has additional benefits, such as ease of product isolation and safety, due to its high heat capacity and unique redox stability.48 Nonpolar solvents, such as toluene and benzene, are not suitable for synthesis of 1,2,3-triazoles with this catalyst (Table 4, Entries 8 and 12). The polar solvents, namely, methanol, DMF, THF, and acetonitrile, yielded the products in moderate yields (Table 4, Entries 9, 11, 13 and 14). However, the DCM yield was very low in comparison to other solvents (Table 4, Entry 10). This is probably due to interference of the solvents with the surface active sites of the catalyst. Interestingly, good yields were observed under solvent-free conditions (Table 4, Entry 6).
With this optimized reaction condition (catalyst, solvent, temperature, and additive-free) in hand, we sought to extend our studies through the synthesis a variety of benzyl halides with sodium azide, followed by [3 + 2] cycloaddition with dissimilar terminal alkynes by using the novel Cu(II) catalyzed process, and the results obtained are shown in Table 5. Likewise, reactions of most of the substrates with several benzyl halides and terminal alkynes bearing electron neutral and electron-donating as well as electron-withdrawing groups were accomplished smoothly and the corresponding 1,2,3-triazoles are attained in good to excellent yields. Note that the benzyl halides generated moderate to excellent yields with aliphatic to aromatic terminal alkynes, respectively. Interestingly, a hetero benzyl halide, namely, 2-picolyl chloride, exhibited an excellent yield with diverse terminal alkynes (Table 5, Entries 3–5). The benzyl halides offered good yields with aromatic terminal alkynes; however, lower yields were observed with aliphatic terminal alkynes (Table 5). The benzyl halide having a strong withdrawing group (–NO2) delivered moderate yields after a long time (Table 5, Entries 13, 17, 22). We also conducted this reaction on a large scale (with benzyl bromide (10 mmol), sodium azide and phenylacetylene as the model reaction) to further advance the industrial utility of the catalytic formulations. Remarkably, we found significant regioselectivity and high yields of the desired products.
| Entry | Halide | Alkyne | Product | Time (h) | Yieldb (%) |
|---|---|---|---|---|---|
| a Reagents and reaction conditions: Halide (1 mmol), sodium azide (1.2 mmol), terminal alkyne (1.1 mmol), catalyst (10 mg), and solvent (1.5 mL).b Yields of isolated products. | |||||
| 1 |  |
 |
 |
4 | 93 |
| 2 |  |
 |
 |
3.5 | 96 |
| 3 |  |
 |
 |
5 | 89 |
| 4 |  |
 |
 |
6 | 91 |
| 5 |  |
 |
 |
6 | 81 |
| 6 |  |
 |
 |
4 | 93 |
| 7 |  |
 |
 |
4 | 94 |
| 8 |  |
 |
 |
6 | 86 |
| 9 |  |
 |
 |
4.5 | 92 |
| 10 |  |
 |
 |
4 | 90 |
| 11 |  |
1-Octyne |  |
7 | 86 |
| 12 |  |
 |
 |
7.5 | 82 |
| 13 |  |
 |
 |
9 | 79 |
| 14 |  |
 |
 |
6 | 88 |
| 15 |  |
 |
 |
7 | 85 |
| 16 |  |
1-Octyne |  |
6 | 83 |
| 17 |  |
 |
 |
8 | 81 |
| 18 |  |
 |
 |
5.5 | 86 |
| 19 | 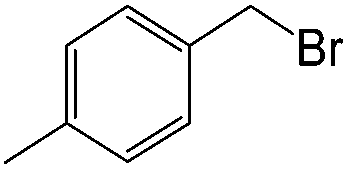 |
 |
 |
7 | 86 |
| 20 |  |
 |
 |
5 | 91 |
| 21 |  |
 |
 |
6 | 90 |
| 22 |  |
1-Octyne |  |
10 | 82 |
| 23 |  |
1-Octyne |  |
9 | 84 |
By analogy with our investigation and earlier reports, the plausible mechanistic pathways for the synthesis of 1,4-disubstituted 1,2,3-triazoles with Cu(II)-PBS-HPMO catalyst are shown in Scheme 3. The one-pot multicomponent click reaction involves first the formation of in situ generated organic azide within a short reaction time and a marginal increase in polarity by benzyl substitution of the azide.29 The role of the Cu species in the porphyrin-bridged PMOs is to facilitate the formation of the Cu(II)–acetylide complex and the activation of the azide function towards nucleophilic attack by reducing the electron density of the alkyne. Finally, the porphyrinatocopper catalyst is regenerated by protonolysis of the intermediate to give the corresponding 1,2,3-triazoles. Interestingly, the used Cu(II)-PBS-HPMO catalyst did not exhibit any significant loss of product yield, and the catalyst could be recycled successively for five runs.
 | ||
| Scheme 3 Plausible reaction mechanism for 1,4-disubstituted 1,2,3-triazole formation with Cu(II)-PBS-HPMO catalyst. | ||
4 Conclusions
In summary, an efficient, straightforward, and atom-economical one-pot synthesis of 1,4-disubstituted 1,2,3-triazoles is reported for the first time using Cu(II)-PBS-HPMO as a heterogeneous catalyst. This simple and environmentally benign catalysis proceeds under mild conditions without any reducing agents or additives. This protocol involved both benzyl halides and hetero benzyl halides, and afforded great yields with terminal alkynes (aromatic and aliphatic). In addition, the Cu(II)-PBS-HPMO showed high reusability with consistent activity for at least five cycles, indicating its potential as a green catalyst; moreover, it has significant potential in large-scale applications.Acknowledgements
ANP thanks the Korean Embassy in India for their support of his stay in South Korea under IKRI Program Fellowship. This work was supported by the National Research Foundation of Korea [NRF] grant funded by the Korea Government [MSIP] [no. 20090083525].References
- (a) M. J. Climent, A. Corma and S. Iborra, Chem. Rev., 2011, 111, 1072 CrossRef CAS PubMed; (b) M. Eissen, J. O. Metzger, E. Schmidt and V. Schneidewind, Angew. Chem., Int. Ed., 2002, 41, 414 CrossRef CAS.
- (a) M. J. Climent, A. Corma and S. Iborra, RSC Adv., 2012, 2, 16 CAS; (b) Y. Gu, Green Chem., 2012, 14, 2091 RSC.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS.
- (a) R. Huisgen in 1,3-Dipolar Cycloadditions. Chem., ed. A. Padwa, Wiley, New York, 1984, vol. 1, p. 1 Search PubMed; (b) A. Padwa in Comprehensive Organic Synthesis, ed. B. M. Trost, Pergamon, Oxford, 1991, vol. 4, p. 1069 Search PubMed.
- Y. Monguchi, K. Nozaki, T. Maejima, Y. Shimoda, Y. Sawama, Y. Kitamura, Y. Kitadeb and H. Sajiki, Green Chem., 2013, 15, 490 RSC.
- S. Löber, P. Rodriguez-Loaiza and P. Gmeiner, Org. Lett., 2003, 5, 1753 CrossRef PubMed.
- K. B. Sharpless and R. Manetsch, Expert Opin. Drug Discovery, 2006, 1, 525 CrossRef CAS PubMed.
- (a) Q. Wang, T. R. Chan, R. Hilgraf, V. V. Fokin, K. B. Sharpless and M. G. Finn, J. Am. Chem. Soc., 2003, 125, 3192 CrossRef CAS PubMed; (b) A. J. Link and D. A. Tirell, J. Am. Chem. Soc., 2003, 125, 11164 CrossRef CAS PubMed; (c) V. G. Nenajdenko, A. V. Gulevich, N. V. Sokolova, A. V. Mironov and E. S. Balenkova, Eur. J. Org. Chem., 2010, 1445 CrossRef CAS.
- (a) D. D. Diaz, K. Rajagopal, E. Strable, J. Schneider and M. G. Finn, J. Am. Chem. Soc., 2006, 128, 6056 CrossRef CAS PubMed; (b) W. B. Zhang, Y. Tu, R. Ranjan, R. M. VanHorn, S. Leng, J. Wang, M. J. Polce, C. Wesdemiotis, R. P. Quirk, G. R. Newkome and S. Z. D. Cheng, Macromolecules, 2008, 41, 515 CrossRef CAS; (c) J. S. Marois, K. Cantin, A. Desmarais and J. F. Morin, Org. Lett., 2008, 10, 33 CrossRef CAS PubMed.
- S. Löber and P. Gmeiner, Tetrahedron, 2004, 60, 8699 CrossRef PubMed.
- J. Gierlich, G. A. Burley, P. M. E. Gramlich, D. M. Hammond and T. Carell, Org. Lett., 2006, 8, 3639 CrossRef CAS PubMed.
- (a) R. Alvarez, S. Valazquez, F. San, S. Aquaro, C. De, C. F. Perno, A. Karlsson, J. Balzarini and M. J. Camarasa, J. Med. Chem., 1994, 37, 4185 CrossRef CAS; (b) S. Velazquez, R. Alvarez, C. Perez, F. Cago, C. De, J. Balzarini and M. J. Camarasa, Antiviral Chem. Chemother., 1998, 9, 481 CAS.
- A. Dondoni and A. Marra, J. Org. Chem., 2006, 71, 7546 CrossRef CAS PubMed.
- E. Fernandez-Megia, J. Correa, I. Rodrguez-Meizoso and R. Riguera, Macromolecules, 2006, 39, 2113 CrossRef CAS.
- Z. Guo, A. Lei, X. Liang and Q. Xu, Chem. Commun., 2006, 4512 RSC.
- A. Dondoni, Chem.–Asian J., 2007, 2, 700 CrossRef CAS PubMed.
- X. C. Cambeiro and M. A. Pericàs, Adv. Synth. Catal., 2011, 353, 113 CrossRef CAS.
- (a) T. R. Chan, R. Hilgraf, K. B. Sharpless and V. V. Fokin, Org. Lett., 2004, 6, 2853 CrossRef CAS PubMed; (b) W. G. Lewis, F. G. Magallon, V. V. Fokin and M. G. Finn, J. Am. Chem. Soc., 2004, 126, 9152 CrossRef CAS PubMed; (c) S. Özçubukçu, E. Ozkal, C. Jimeno and M. A. Pericàs, Org. Lett., 2009, 11, 4680 CrossRef PubMed.
- R. J. Detz, S. A. Heras, R. de Gelder, P. W. N. M. van Leeuwen, H. Hiemstra, J. N. H. Reek and J. H. van Maarseveen, Org. Lett., 2006, 8, 3227 CrossRef CAS PubMed.
- E. Hao, Z. Wang, L. Jiao and S. Wang, Dalton Trans., 2010, 39, 2660 RSC.
- (a) P. K. Kadaba, J. Med. Chem., 1988, 31, 196 CrossRef CAS; (b) W. Junmin, J. Changsoo, C. Kyuyun, K. Kyungchell and Q. Zeshan, Prog. Nat. Sci., 2006, 16, 925 CrossRef; (c) J. L. Kelley, C. S. Koble, R. G. Davis, E. W. Mclean, F. E. Soroko and B. R. Cooper, J. Med. Chem., 1995, 38, 4131 CrossRef CAS.
- (a) H. C. Kolb and K. B. Sharpless, Drug Discovery Today, 2003, 8, 1128 CrossRef CAS; (b) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef PubMed.
- P. Appukkuttan, W. Dehaen, V. V. Fokin and E. van der Eycken, Org. Lett., 2004, 6, 4223 CrossRef CAS PubMed.
- (a) F. Himo, T. Lovell, R. Hilgraf, V. V. Rostovtsev, L. Noodleman, K. B. Sharpless and V. V. Fokin, J. Am. Chem. Soc., 2005, 127, 210 CrossRef CAS PubMed; (b) H. A. Orgueira, D. Fokas, Y. Isome, P. C.-M. Chan and C. M. Baldino, Tetrahedron Lett., 2005, 46, 2911 CrossRef CAS PubMed.
- F. Wang, H. Fu, Y. Jiang and Y. Zhao, Green Chem., 2008, 10, 452 RSC.
- D. M. Haddleton, C. B. Jasieczek, M. J. Hannon and A. J. Shooter, Macromolecules, 1997, 30, 2190 CrossRef CAS.
- (a) V. O. Rodionov, S. I. Presolski, S. Gardinier, Y.-H. Lim and M. G. Finn, J. Am. Chem. Soc., 2007, 129, 12696 CrossRef CAS PubMed; (b) V. O. Rodionov, S. I. Presolski, D. D. Diaz, V. V. Fokin and M. G. Finn, J. Am. Chem. Soc., 2007, 129, 12705 CrossRef CAS PubMed.
- M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952 CrossRef CAS PubMed.
- (a) T. Miao and L. Wang, Synthesis, 2008, 363 CAS; (b) T. Shamin and S. Paul, Catal. Lett., 2010, 136, 260 CrossRef PubMed.
- B. S. Lee, M. Yi, S. Y. Chu, J. Y. Lee, R. Kwon, K. R. Lee, D. Kang, W. S. Kim, H. B. Lim, J. Lee, H.-J. Youn, D. Y. Chi and N. H. Hur, Chem. Commun., 2010, 46, 3935 RSC.
- (a) M. Lammens, J. Skey, S. Wallyn, R. O'Reilly and F. D. Prez, Chem. Commun., 2010, 46, 8719 RSC; (b) Y. M. A. Yamada, S. M. Sarkar and Y. Uozumi, J. Am. Chem. Soc., 2012, 134, 9285 CrossRef CAS PubMed.
- B. H. Lipshutz and B. R. Taft, Angew. Chem., Int. Ed., 2006, 45, 8235 CrossRef CAS PubMed.
- A. Kumar, S. Aerry, A. Saxena, A. De and S. Mozumdar, Green Chem., 2012, 14, 1298 RSC.
- (a) S. Chassaing, A. Alix, T. Boningari, K. S. S. Sido, M. Keller, P. Kuhn, B. Louis, J. Sommer and P. Pale, Synthesis, 2010, 1557 CAS; (b) V. Bénéteau, A. Olmos, T. Boningari, J. Sommer and P. Pale, Tetrahedron Lett., 2010, 51, 3673 CrossRef PubMed.
- (a) K. Namitharan, M. Kumarraja and K. Pitchumani, Chem.–Eur. J., 2009, 15, 2755 CrossRef CAS PubMed; (b) A. N. Prasad, B. Thirupathi, G. Raju, R. Srinivas and B. M. Reddy, Catal. Sci. Technol., 2012, 2, 1264 RSC.
- I. S. Park, M. S. Kwon, Y. Kim, J. S. Lee and J. Park, Org. Lett., 2008, 10, 497 CrossRef CAS PubMed.
- M. Waki, N. Mizoshita, T. Ohsuna, T. Tani and S. Inagaki, Chem. Commun., 2010, 46, 8163 RSC.
- X. Zheng, M. Wang, Z. Sun, C. Chen, J. Ma and J. Xu, Catal. Commun., 2012, 29, 149 CrossRef CAS PubMed.
- X. Qiu, S. Han and M. Gao, J. Mater. Chem. A, 2013, 1, 1319 CAS.
- E.-Y. Jeong, M. B. Ansari and S.-E. Park, ACS Catal., 2011, 1, 855 CrossRef CAS.
- A. Modak, J. Mondal and A. Bhaumik, Green Chem., 2012, 14, 2840 RSC.
- (a) E.-Y. Jeong, A. Burri, S.-Y. Lee and S.-E. Park, J. Mater. Chem., 2010, 20, 10869 RSC; (b) C. E. Diaz-Uribe, M. C. Daza, F. Martínez, E. A. Páez-Mozo, C. L. B. Guedes and E. Di Mauro, J. Photochem. Photobiol., A, 2010, 215, 172 CrossRef CAS PubMed; (c) H. Peng and Y. Lu, Adv. Mater., 2008, 20, 797 CrossRef CAS.
- D. F. Marsh and L. M. Mink, J. Chem. Educ., 1996, 73, 1188 CrossRef CAS.
- B. C. Barja and P. F. Aramendía, Photochem. Photobiol. Sci., 2008, 7, 1391 CAS.
- (a) A. N. Prasad, R. Srinivas and B. M. Reddy, Catal. Sci. Technol., 2013, 3, 654 RSC; (b) M. K. Patil, A. N. Prasad and B. M. Reddy, Curr. Org. Chem., 2011, 15, 3961 CrossRef CAS; (c) A. N. Prasad and B. M. Reddy, Curr. Catal., 2013, 2, 159 CrossRef CAS; (d) B. M. Reddy and M. K. Patil, Chem. Rev., 2009, 109, 2185 CrossRef CAS PubMed.
- B. Thirupathi, R. Srinivas, A. N. Prasad, J. K. P. Kumar and B. M. Reddy, Org. Process Res. Dev., 2010, 14, 1457 CrossRef CAS.
- S. Narayan, J. Muldoon, M. G. Finn, V. V. Fokin, H. C. Kolb and K. B. Sharpless, Angew. Chem., Int. Ed., 2005, 44, 3275 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR and 13C NMR data and spectras of isolated compounds. See DOI: 10.1039/c4ra04093g |
| This journal is © The Royal Society of Chemistry 2014 |