
Inflammatory modulation of stem cells by Magnetic Resonance Imaging (MRI)-detectable nanoparticles†
Abstract
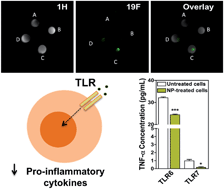
In the current work, we labelled human hematopoietic stem cells with polymeric nanoparticles (NPs) that can be tracked by Magnetic Resonance Imaging (MRI) and studied their effect on cell metabolism, proliferation, secretomics, genomics and differentiation. We showed that NPs had no effect on the stem cell differentiation program but affected their paracrine activity.
 Please wait while we load your content...
Please wait while we load your content...

