Hydrogen bond donor–acceptor–donor organocatalysis for conjugate addition of benzylidene barbiturates via complementary DAD–ADA hydrogen bonding†
Franco King-Chi Leung,
Jian-Fang Cui,
Tsz-Wai Hui,
Zhong-Yuan Zhou and
Man-Kin Wong*
State Key Laboratory of Chirosciences and Department of Applied Biology and Chemical Technology, The Hong Kong Polytechnic University, Hung Hom, Kowloon, Hong Kong, China. E-mail: mankin.wong@polyu.edu.hk; Fax: +852 3400-8701
First published on 10th June 2014
Abstract
A new class of hydrogen bond donor–acceptor–donor (HB-DAD) organocatalysts has been developed for conjugate addition of benzylidene barbiturates. HB-DAD organocatalyst 1a (featuring para-chloro-pyrimidine as the hydrogen bond acceptor (HBA), N–H as the hydrogen bond donor (HBD) and a trifluoroacetyl group as the electron withdrawing group (EWG)) is able to activate benzylidene barbiturates through complementary DAD–ADA hydrogen bonding. Using 1a in benzylidene barbiturate conjugate addition, good yields were achieved. The relative rate constant (krel = 2.9) of 1a in catalyzing the conjugate addition of benzylidene barbiturates and the binding constant (KA = 8936 (±723) M−1) of 1a with benzylidene barbiturates were determined by NMR and UV/Vis. spectroscopy studies. The excellent correlation (R2 = 0.97) between the relative rate constant and binding affinity of 1a with benzylidene barbiturates provides support for the importance of DAD–ADA hydrogen bonding in organocatalysis.
Introduction
Hydrogen bond donor–donor (HB-DD) organocatalysis has largely been developed as efficient methodologies to achieve synthetic organic transformations.1 Over time, significant advancements have been made in HB-DD organocatalysis employing thiourea-,2 guanidinium-3 and squaramide-based4 HB-DD organocatalysts. Given the success of the HB-DD organocatalysis, it remains a great interest to explore new catalyst scaffolds for hydrogen bond-based organocatalysis.
Hydrogen bond donor–acceptor–donor (HB-DAD) and hydrogen bond acceptor–donor–acceptor (HB-ADA) systems are common in supramolecular chemistry, mainly acting as supramolecular linking units in non-covalent polymer assembly.5 This class of hydrogen bonding system is of high utility in various applications in materials science because of its highly directional nature. In addition, these three complementary DAD–ADA hydrogen bondings are strong binding arrays.6 However, studies on the use of the complementary DAD–ADA hydrogen bonding in organocatalysis have rarely been explored.
Along with our ongoing interest in the development of organocatalysis for organic synthesis7 and bioconjugation,8 we envision that this highly directional and strong complementary HB-DAD and HB-ADA systems could be developed as new catalyst scaffolds and efficient activation modes for hydrogen bond-based organocatalysis.
In this work, we have designed HB-DAD organocatalysts consisting of three components (1) hydrogen bond acceptor (HBA), (2) hydrogen bond donor (HBD), and (3) electron withdrawing group (EWG) (Scheme 1). The nitrogen atom in N-heterocyclic aromatic rings including chloro-pyrimidine, pyridine, and pyrazine were selected as HBA to give structurally diverse catalyst scaffolds. Nitrogen–hydrogen (N–H bond), one of the most electronegative hydrogen bonds, was chosen as HBD in the design. The electron withdrawing group could be used to tune the electrophilicity of the N–H bond.
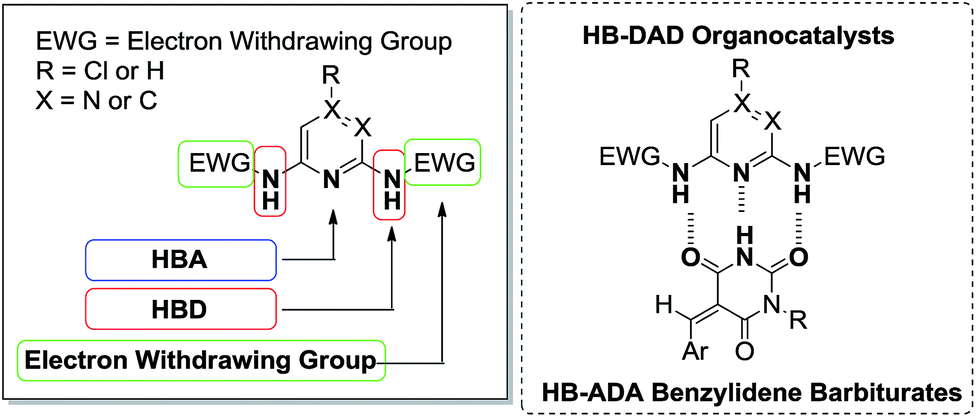 | ||
| Scheme 1 Design of HB-DAD organocatalysts containing (1) HBA, (2) HBD, and (3) electron withdrawing group. Complementary DAD–ADA hydrogen bonding between HB-DAD organocatalysts and HB-ADA benzylidene barbiturates. | ||
Benzylidene barbiturates are biologically active compounds9 and synthetic building blocks.10 We considered benzylidene barbiturates as HB-ADA substrates because of (1) the imide group functioning as HB-ADA moiety and (2) the electron deficient alkene unit amenable for nucleophilic attack. The complementary hydrogen bonding mode of HB-DAD organocatalysts and HB-ADA benzylidene barbiturates is depicted in Scheme 1.
In 2011, Spange and co-workers found that a gradual adjustment of electrophilicity of a barbiturate merocyanine is achieved through cooperative DAD–ADA hydrogen bond.11a Substituent effects of HB-DAD receptors are transmitted to the reactive center of electrophilic HB-ADA substrates so that a fine adjustment of their reactivity would be possible.11b,c
In the present work, we developed a organocatalytic conjugate addition of benzylidene barbiturates with 2-methylfuran catalyzed by HB-DAD organocatalysts through the complementary DAD–ADA hydrogen bonding. Kinetic studies of HB-DAD organocatalysts in catalyzing conjugate addition of benzylidene barbiturates and binding constant studies of HB-DAD organocatalysts with HB-ADA benzylidene barbiturates were conducted. The excellent correlation between the binding constants and relative rate constants of HB-DAD organocatalysts provides support for HB-DAD as the catalyst scaffold in organocatalysis.
Results and discussion
Preparation of HB-DAD organocatalysts 1–4 and HB-ADA benzylidene barbiturates 5 and 7
HB-DAD organocatalysts 1a–c and 3a–c were prepared by amide coupling of 2,6-diamino-4-chloropyrimidine/2,6-diaminopyrazine with acid chlorides/anhydrides and obtained in 18–68% isolated yield. In addition, 1a was characterized by X-ray crystallography (see ESI†). HB-DAD organocatalysts 2a,12 2b,13 2c,14 4a,15 4b, and 4c16 were synthesized according to literature reports. HB-ADA benzylidene barbiturates 5 and 7 were prepared by condensation of barbiturate acid derivatives with various benzaldehydes and obtained in 24–95% isolated yield. The E/Z ratio of the alkene moieties of 5 and 7 were found to be 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by NMR studies.
:1 by NMR studies.
Catalytic activities of HB-DAD organocatalysts 1–3 in conjugate addition of benzylidene barbiturates
As shown in Scheme 2, the catalytic activity of 20 mol% of 1a in conjugate addition of benzylidene barbiturate 5a (0.05 mmol) and 2-methylfuran (0.05 mmol) at 25 °C in 24 h were investigated. Adduct 6a was obtained in 61% yield, using toluene as internal standard determined by 1H NMR studies. In the absence of 1a, 6a was obtained in 28% yield. | ||
| Scheme 2 Catalytic activities of HB-DAD organocatalysts 1–3 in conjugate addition of 5a. | ||
To optimize the reaction conditions of the conjugate addition of benzylidene barbiturate 5a with 2-methylfuran, reaction temperature, choice of solvents, and amount of HB-DAD organocatalyst 1a used were studied (see ESI†). The conjugate addition in the presence of 20 mol% of 1a in CH2Cl2 at 25 °C in 24 h was found to be the optimized reaction conditions. The catalytic activities of a variety of HB-DAD organocatalysts 1–3 towards conjugate addition of benzylidene barbiturate 5a were examined accordingly (Scheme 2).
Using 1a with trifluoroacetyl group as the EWG, adduct 6a was obtained in 61% yield. Yet, 40% yield of 6a was obtained using 1b (bearing hexanoyl group as the EWG). The results indicated that the electron withdrawing trifluoroacetyl group is important to achieve high catalytic activity.17 1c with pivaloyl group as the EWG gave no enhancement to yield of 6a. These findings indicated that the steric effect of the pivaloyl group would lead to poor catalytic activity in the reaction. Hence, the activating effect of the EWG on the catalytic activities of HB-DAD organocatalysts is in the order of 1a-trifluoroacetyl group > 1b-hexanoyl group > 1c-pivaloyl group. Interestingly, this trend of activating effect of the EWG on the catalytic activities of 1 also applies for HB-DAD organocatalysts 2 and 3.
Using 1a with chloro-pyrimidine as the HBA, adduct 6a was obtained in 61% yield while 2a with pyridine as the HBA gave 49% yield of 6a. The reaction using 3a with pyrazine as the HBA gave only 44% yield of 6a. These results indicated that the activating effect of HBA on catalytic activities of HB-DAD organocatalysts is in the order of 1a-chloro-pyrimidine > 2a-pyridine > 3a-pyrazine.
The importance of hydrogen bond donors and acceptors in HB-DAD organocatalyst scaffolds
To investigate the importance of hydrogen bond donors and acceptors of HB-DAD organocatalysts in catalyzing the conjugate addition, hydrogen bond organocatalysts including D–D class 4a, DA- class 4b and D--class 4c were employed for conjugate addition of benzylidene barbiturate 5a by 2-methylfuran.
Using 4a, adduct 6a was obtained in 28% yield. In contrast, the reaction using 2a gave adduct 6a in 49% yield (Scheme 3). Note that 2a has a nitrogen atom yet 4a bears a C–H bond. Thus, the nitrogen atom (HBA) of 2a in the DAD–ADA hydrogen bonding is essential to give catalytic activities on conjugate addition of 5a.
 | ||
| Scheme 3 2a and 4a in catalyzing conjugate addition of 5a. | ||
Conjugate addition of 5a using DA- class 4b (bearing one trifluoroacetamide group) and nitrogen atom (HBA) and D--class 4c (bearing one trifluoroacetamide group) gave adduct 6a in 28% yield (Scheme 4). As a comparison, 2a (bearing HB-DAD catalyst scaffold) gave 49% yield. The higher yield of 2a than 4b and 4c indicated that the trifluoroacetamide group and nitrogen atom are important for the catalysis.
 | ||
| Scheme 4 2a, 4b and 4c in catalyzing conjugate addition of 5a. | ||
Using HB-DD organocatalyst thiourea A (3,5-bis(trifluoromethyl)phenyl thiourea),18 adduct 6a was obtained in 64% yield (Scheme 5) while using HB-DAD organocatalyst 1a gave 61%. The results indicated that 1a afforded comparable catalytic activity to thiourea A in conjugate addition of 5a.
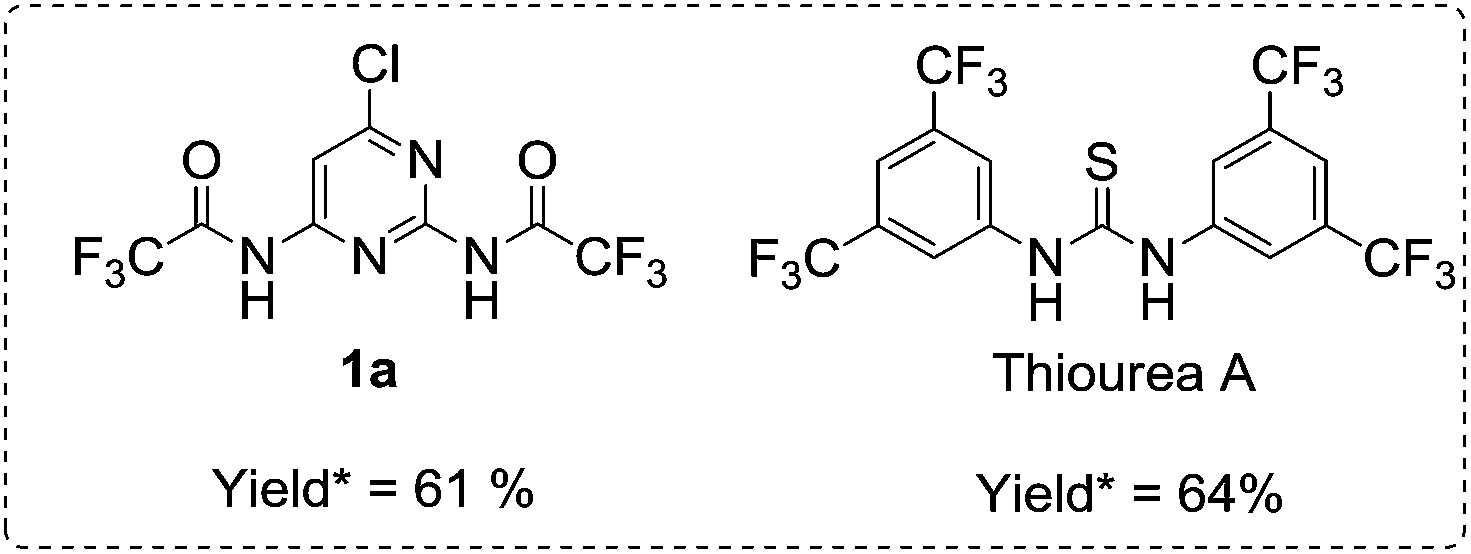 | ||
| Scheme 5 1a and thiourea A in catalyzing conjugate addition of 5a. | ||
Substrate scope of conjugate addition of benzylidene barbiturates 5 and 7
The substrate scope of conjugate addition was examined by using a variety of benzylidene barbiturates. Treatment of a series of benzylidene barbiturates 5a–o with 2-methylfuran furnished the corresponding adducts 6a–o (Table 1). As shown, the conjugate additions were conducted in the presence of 20 mol% of 1a at 25 °C in 24 h. The 1a-catalyzed conjugate addition worked well for electron rich benzylidene barbiturates 5a–h with good yield (Table 1; entries 1–8) because of the low background NMR yield of the reactions. Particularly, 1a-catalyzed conjugate addition of 5b and 5e–5h bearing para-alkoxy phenyl groups afforded good yield (entries 2 and 5–8) while the conjugate addition of 5d bearing an ortho-methoxy phenyl group could give even higher yield (entry 4). In contrast, 5c bearing a meta-methoxy phenyl group gave a higher value of background NMR yield (65%), probably due to the methoxy group in the meta-position contributes less positive mesomeric effect. The results indicated that benzylidene barbiturates bearing electron donating groups could lead to better yield because of the lower background yield.
|
||||||
|---|---|---|---|---|---|---|
| Entrya | Substrate | Ar | Product | Isolated yield (%) | NMR yieldb (%) | Background NMR yieldb,c (%) |
| a Reaction conditions: 5a–5o (0.05 mmol), 2-methylfuran (0.05 mmol), 1a (0.01 mmol), CH2Cl2 (1 mL), 25 °C, 24 h.b Yields were determined by 1H NMR of the crude product using toluene as the internal standard.c Without addition of 1a. | ||||||
| 1 | 5a |  |
6a | 55 | 61 | 28 |
| 2 | 5b |  |
6b | 52 | 57 | 25 |
| 3 | 5c | 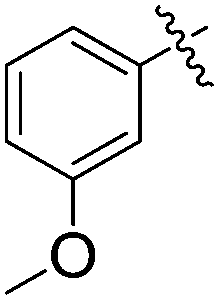 |
6c | 78 | 80 | 65 |
| 4 | 5d |  |
6d | 25 | 22 | 7 |
| 5 | 5e |  |
6e | 30 | 30 | 17 |
| 6 | 5f |  |
6f | 16 | 20 | 7 |
| 7 | 5g |  |
6g | 65 | 70 | 31 |
| 8 | 5h | 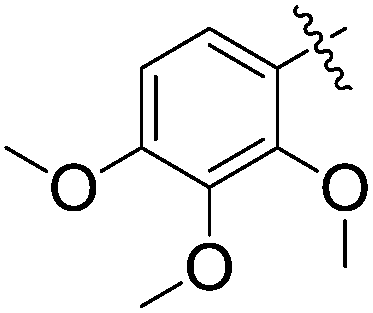 |
6h | 47 | 50 | 21 |
| 9 | 5i |  |
6i | 66 | 69 | 30 |
| 10 | 5j |  |
6j | 73 | 71 | 36 |
| 11 | 5k |  |
6k | 79 | 84 | 60 |
| 12 | 5l |  |
6l | 35 | 38 | 16 |
| 13 | 5m |  |
6m | 95 | 96 | 87 |
| 14 | 5n |  |
6n | 95 | 97 | 76 |
| 15 | 5o |  |
6o | 98 | 99 | 99 |

Interestingly, 5i bearing an isopropyl phenyl group, 5j bearing a t-butyl phenyl group and 5l bearing a napthalene group led to the corresponding adducts in good yield (entries 9,10 and 12) and low background NMR yield (16–36%). However, 5k bearing a phenyl group afforded 60% background NMR yield (entry 11). Note that the alkyl- and aryl-substituents on the phenyl ring of benzylidene barbiturates have positive inductive and mesomeric effect on the phenyl rings. In this connection, benzylidene barbiturates bearing electron donating groups led to better yield with low background yield.
Conjugate additions of 5m–o bearing electron deficient substituents –Cl, –Br and –CN gave high background NMR yield (76–99%) (entries 13–15). These findings indicated that the electrophilicity of benzylidene barbiturates was a crucial factor in governing the yield in the reaction.
We further examined the scope of this reaction by changing the substituents on the barbiturate acid moiety of benzylidene barbiturates 7a–d to give the corresponding adducts 8a–d (Table 2). Benzylidene barbiturates 7a and 7b (R = methyl) afforded good yield (Table 2, entries 1 and 2). In contrast, conjugate additions of 7c and 7d (R = m-tolyl) gave high background NMR yield (27–61%; entries 3 and 4). The difference in the background NMR yield was possibly due to the increased electrophilicity of benzylidene barbiturates (7c and 7d) (i.e., m-tolyl group giving negative mesomeric effect on the benzylidene barbiturates).
|
|||||||
|---|---|---|---|---|---|---|---|
| Entrya | Substrate | R | Ar | Product | Isolated yield (%) | NMR yieldb (%) | Background NMR yieldb,c (%) |
| a Reaction conditions: 7a–7d (0.05 mmol), 2-methylfuran (0.05 mmol), 1a (0.01 mmol), CH2Cl2 (1 mL), 25 °C, 24 h.b Yields were determined by 1H NMR of the crude product using toluene or ethyl acetate as the internal standard.c Without addition of 1a. | |||||||
| 1 | 7a | CH3 |  |
8a | 20 | 25 | 5 |
| 2 | 7b | CH3 |  |
8b | 20 | 18 | 6 |
| 3 | 7c |  |
 |
8c | 46 | 48 | 27 |
| 4 | 7d |  |
 |
8d | 75 | 73 | 61 |

Furthermore, we examined the scope of nucleophiles (1-methylindole, indole, 5-methoxylindole, thiophene, dibenzoylmethane and ethylbenzyolacetate) in 1a- and thiourea A-catalyzed conjugate additions of 5a. However, no yield increase of the reaction was observed using 1a or thiourea A (Scheme 6 and Table S4 and S5†). In this regard, 1a and thiourea A have shown similar behavior in catalyzing conjugate addition of 5a.
 | ||
| Scheme 6 1a and thiourea A in catalyzing conjugate addition of 5a with nucleophiles. | ||
Time course experiments using 20 mol% of HB-DAD organocatalysts 1a, 1b, 1c, 2a, 2b, and 3a in conjugate additions of 5a (0.025 mmol; 0.05 M) with 2-methylfuran (0.25 mmol; 0.5 M) to give adduct 6a in CDCl3 at 25 °C in 120 min were monitored by 1H NMR.19,20 As shown in Fig. 1, the reaction orders were nearly constant over time indicating the absence of product inhibition. Using 1a gave adduct 6a in 66% yield while using 2a gave adduct 6a in 56% yield. Both 1b and 2b were found to be catalytically active, giving 6a in 52% and 49% yields, respectively. In addition, the conjugate addition using 3a could lead to adduct 6a in 44% yield. However, 1c was found to be inactive to catalyze conjugate addition of 5a with 2-methylfuran (yield = 22%). With reference to the increasing acidity of the HBD (N–H) moieties of HB-DAD organocatalysts, the reaction rate is in the order of 1a > 2a > 1b > 2b > 3a.
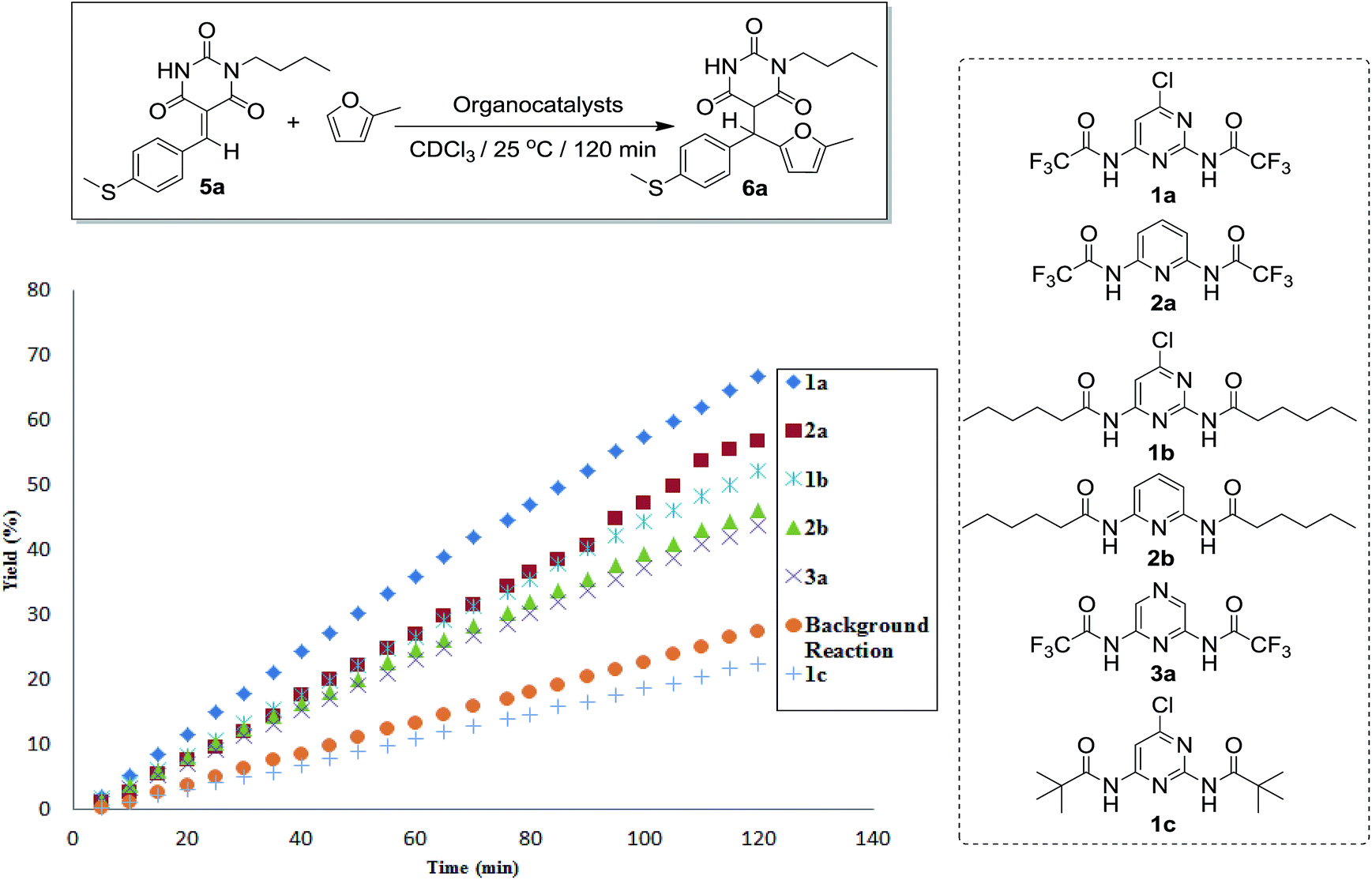 | ||
| Fig. 1 Time course experiments of HB-DAD organocatalyst-catalyzed conjugate addition of 5a with 2-methylfuran. | ||
Kinetic studies of HB-DAD organocatalysts
With a 10-fold excess of 2-methylfuran, all the conjugate additions of 5a were regarded as pseudo-first-order, and the corresponding rate constant kobs were determined and depicted in Table 3. For the determination of kobs, the kinetics data was plotted as ln[5a] against the reaction time.19 The rate constant kobs was determined by the negative slope of the plot (see ESI†).| Entry | Catalyst | kobs × 10−4 (s−1) | kcat × 10−4 (s−1) | krel |
|---|---|---|---|---|
| 1 | 1a | 1.48 | 1.11 | 2.9 |
| 2 | 1b | 1.05 | 0.67 | 1.8 |
| 3 | 1c | 0.37 | −0.01 | −0.03 |
| 4 | 2a | 1.22 | 0.84 | 2.2 |
| 5 | 2b | 0.90 | 0.52 | 1.4 |
| 6 | 3a | 0.82 | 0.44 | 1.2 |
| 7 | — | 0.38 | kuncata = 0.38 | — |
On the basis of the rate constant (kobs), the relative rate constant (krel) of HB-DAD organocatalysts were calculated.20,21 The relative rate constant (krel) of 1a-catalyzed conjugate addition of 5a was 2.9 (Table 3; entry 1). The relative rate constant suggests that 20 mol% of HB-DAD organocatalyst 1a increases the conjugate addition rate by a factor of = 2.9. The krel of 2a was 2.2 (entry 4) while the krel of 3a was calculated as 1.2 (entry 6). In addition, the krel of 1b and 2b were 1.8 and 1.4, respectively (entries 2 and 5). The krel of 1c was −0.03 (entry 3), meaning that 20 mol% of 1c gave no catalytic activity to conjugate addition of 5a with 2-methylfuran. These results indicated that the more electron deficient EWG (trifluoroacetyl group) afforded the higher catalytic activities than using the hexanoyl group as the EWG.
Binding studies of HB-DAD organocatalysts
To quantify the binding affinity of HB-DAD organocatalysts and HB-ADA benzylidene barbiturate derivatives, binding constant studies were employed. In these studies, binding constants were monitored with UV/Vis. spectroscopy titration experiments.11,22The linearized Scatchard plot was used in determining the binding constants of 1a, 1b, 2a, and 2b with barbiturate 9 (see ESI†). The binding constant (KA = 8936 (±723) M−1; Table 4, entry 1) was obtained for 1a. These results indicated that using the trifluoroacetyl group as the EWG led to a significant increase in the binding affinity. Notably, chloro-pyrimidine is a better HBA than pyridine in achieving high binding affinity. With the increasing acidity of HBD (N–H) moieties in HB-DAD organocatalysts, the stability of hydrogen bonding complexes is in the order of 1a > 2a > 1b > 2b.
|
||
|---|---|---|
| Entry | Catalyst | KA (M−1) |
| 1 | 1a | 8936 (±723) |
| 2 | 1b | 6447 (±380) |
| 3 | 2a | 7747 (±367) |
| 4 | 2b | 4895 (±1019) |

Correlation of rate constants and binding constants of HB-DAD organocatalysts
A gradually escalating trend of relative rate constants of conjugate addition of 5a was obtained in kinetic studies while increasing trend in binding affinity was also determined in binding studies. Particularly, a correlation was observed between kinetic and binding studies. The natural logarithm of the binding constants and relative rate constants were listed in Table S8 (see ESI†).20b By plotting of lnKA against lnkrel of HB-DAD organocatalysts, a linear correlation (R2 = 0.97) was obtained (Fig. 2). The results indicated that a higher binding constant gave a higher relative rate of the conjugate addition.
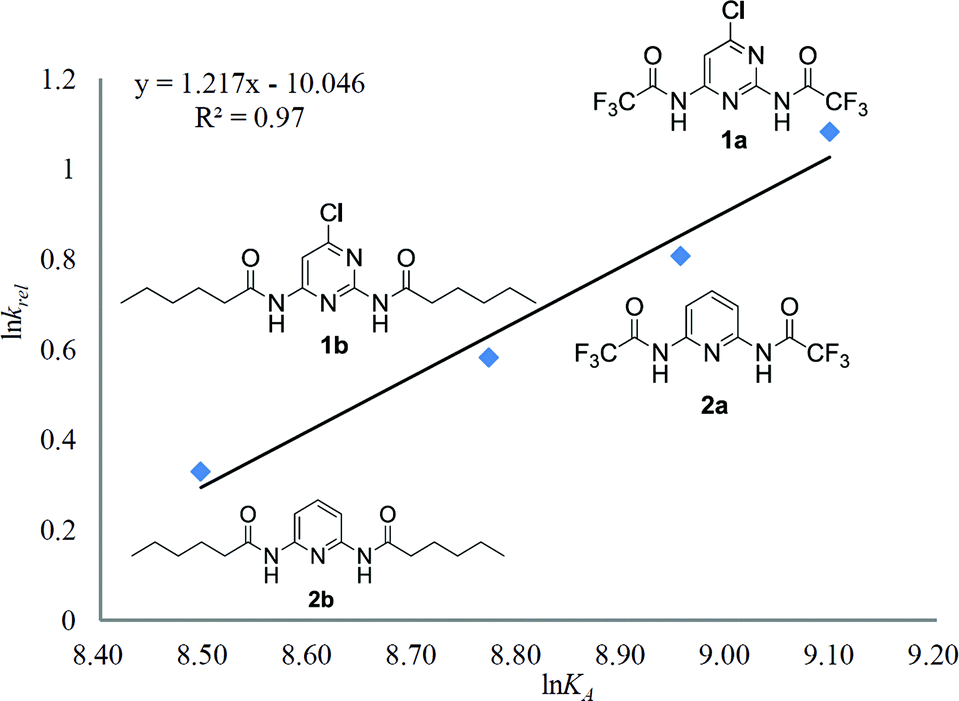 | ||
| Fig. 2 Correlation of natural logarithm rate constants and binding constants of HB-DAD organocatalysts. | ||
Conclusion
In summary, we have developed new hydrogen bond-based organocatalysis using HB-DAD catalyst scaffold in catalyzing the conjugate addition of benzylidene barbiturates. The catalytic activities of HB-DAD organocatalyst 1a were comparable to thiourea A. The catalytic activities of the HB-DAD catalyst scaffolds were supported by the correlation of rate constants and binding constants. This work would lay down a foundation for the development of chiral HB-DAD organocatalysts for asymmetric catalysis.Experimental section
General procedure for synthesis of HB-DAD organocatalysts 1a and 3a
A mixture of 2,6-diamino-N-heterocyclic compounds (1.0 mmol) and trifluoroacetic anhydride (3.0 mmol) in CH2Cl2 (10 mL) was stirred under nitrogen atmosphere at room temperature for 24 h. The reaction mixture was added with water (5 mL) and extracted with ethyl acetate (3 × 10 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated. The residue was purified by flash column chromatography on silica gel using ethyl acetate–hexane as eluent to give 1a (58% yield) and 3a (68% yield).General procedure for synthesis of HB-DAD organocatalysts 1b, 1c, 3b and 3c
A mixture of 2,6-diamino-N-heterocyclic compounds (1.0 mmol), acid chloride (2.5 mmol), 4-dimethylaminopyridine (0.2 mmol) and triethylamine (2.5 mmol) in CH2Cl2 (10 mL) was stirred under nitrogen atmosphere at room temperature for 24 h. The reaction mixture was treated with water (5 mL) and extracted with ethyl acetate (3 × 10 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated. The residue was purified by flash column chromatography on silica gel using ethyl acetate–hexane as eluent to give 1b (15% yield), 1c (62% yield), 3b (58% yield) and 3c (58% yield).General procedure for synthesis of benzylidene barbiturates
A mixture of barbiturate acid (2.0 mmol) and benzaldehyde (2.0 mmol) in EtOH (10 mL) was refluxed for 2–12 h. The reaction mixture was allowed to cool to room temperature and filtered to obtain solid/crystalline crude products. The residue was purified by flash column chromatography on silica gel using ethyl acetate–hexane as eluent to give benzylidene barbiturates in 24–95% yield.Procedure for catalytic conjugate additions of benzylidene barbiturates
A mixture of benzylidene barbiturates 5 (0.05 mmol), 2-methylfuran (0.05 mmol) and HB-DAD organocatalyst 1a (0.01 mmol) in CH2Cl2 (1 mL) was stirred at 25 °C for 24 h. The product yield was determined by crude 1H NMR with toluene (0.02 mmol) as internal standard. The reaction mixture was concentrated. The residue was purified by flash column chromatography on silica gel using ethyl acetate–hexane as eluent to obtain isolated yield.Kinetics study
All reactions were conducted with 0.025 mmol of benzylidene barbiturate 5a, 0.25 mmol of 2-methylfuran, 0.03 mmol of dichloromethane (internal standard) and 20 mol% of HB-DAD organocatalysts. Stock solutions of 5a (0.083 M; 0.05 mmol of 5a in 0.6 mL of CDCl3) and the HB-DAD organocatalysts (0.05 M; 0.01 mmol of HB-DAD organocatalysts in 0.2 mL of CDCl3) were prepared in 2 mL vials. A NMR tube was charged with 0.3 mL of 5a stock solution followed by 0.1 mL of HB-DAD organocatalysts stock solution and 1.98 μL of dichloromethane. The mixture was made up to 0.5 mL with CDCl3. After adding 22.3 μL of 2-methylfuran, the first NMR spectrum was taken 5 min after the addition. Additional NMR spectra were recorded every 5 min for a total of 120 min.Binding study
Ten graduated flasks (5 mL) were added with 0.5 mL of stock solution of barbiturate 9 (2 × 10−4 M; 2 × 10−2 mmol of barbiturate 9 in 100.0 mL of CH2Cl2) (final concentration: 2 × 10−5 M) and 0, 20, 40, 80, 160, 320, 640, 1260, 2560, 3000 μL (corresponding to a 2–290 fold excess) of a stock solution of HB-DAD organocatalysts (9.67 × 10−3 M; 9.67 × 10−2 mmol of HB-DAD organocatalysts in 10.0 mL of CH2Cl2), and filled up to 5 mL with CH2Cl2. The change in absorbance was monitored and evaluated by linearized Scatchard plot. The given values of KA were the average of two runs.Acknowledgements
We thank the financial support of Hong Kong Research Grants Council (PolyU 5031/11P). The Hong Kong Polytechnic University (G-YM50). We also thank Dr Michael Fuk-Yee Kwong and Ms Pui-Ying Choy of PolyU for providing some benzaldehyde substrates.Notes and references
- (a) A. Berkessel and H. Groeger, Asymmetric Organocatalysis: from Biomimetic Concepts to Applications in Asymmetric Synthesis, University Science Books, Mill Valley, 2005 Search PubMed; (b) A. G. Doyle and E. N. Jacobsen, Chem. Rev., 2007, 107, 5713 CrossRef CAS; (c) X. Yu and W. Wang, Chem.–Asian J., 2008, 3, 516 CrossRef CAS PubMed; (d) Z. Zhang and P. R. Schreiner, Chem. Soc. Rev., 2009, 38, 1187 RSC; (e) D. Leow and C. H. Tan, Chem.–Asian. J., 2009, 4, 488 CrossRef CAS PubMed; (f) E. N. Jacobsen and D. W. C. MacMillan, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20618 CrossRef CAS PubMed.
- (a) M. S. Sigman and E. N. Jacobsen, J. Am. Chem. Soc., 1998, 120, 4901 CrossRef CAS; (b) T. P. Yoon and E. N. Jacobsen, Angew. Chem., Int. Ed., 2005, 44, 466 CrossRef CAS; (c) Y. Hoashi, T. Okino and Y. Takemoto, Angew. Chem., Int. Ed., 2005, 44, 4032 CrossRef CAS; (d) T. Inokuma, Y. Hoashi and Y. Takemoto, J. Am. Chem. Soc., 2006, 128, 9413 CrossRef CAS; (e) D. A. Yalalov, S. B. Tsogoeva, T. E. Shubina, I. M. Martynova and T. Clark, Angew. Chem., Int. Ed., 2008, 47, 6624 CrossRef CAS; (f) K. L. Kimmel, M. T. Robak and J. A. Ellman, J. Am. Chem. Soc., 2009, 131, 8754 CrossRef CAS; (g) W. Yang and D. M. Du, Org. Lett., 2010, 12, 5450 CrossRef CAS; (h) K. Asano and S. Matsubara, J. Am. Chem. Soc., 2011, 133, 16711 CrossRef CAS; (i) J. Xu, Y. Hu, D. Huang, K. Wang, C. Xu and T. Niu, Adv. Synth. Catal., 2012, 354, 515–526 CrossRef CAS; (j) K. Bera and I. N. N. Namboothiri, Adv. Synth. Catal., 2013, 355, 1265 CrossRef CAS; (k) J. C. Anderson and P. J. Koovits, Chem. Sci., 2013, 4, 2897 RSC; (l) A. R. Brown, C. Uyeda, C. A. Brotherton and E. N. Jacobsen, J. Am. Chem. Soc., 2013, 135, 6747 CrossRef CAS; (m) M. P. Lalonde, M. A. McGowan, N. S. Rajapaksa and E. N. Jacobsen, J. Am. Chem. Soc., 2013, 135, 1891 CrossRef; (n) H. N. Yuan, S. Wang, J. Nie, W. Meng, Q. Yao and J. A. Ma, Angew. Chem., Int. Ed., 2013, 52, 3869 CrossRef CAS; (o) H. Y. Bae, J. H. Sim, J. W. Lee, B. List and C. E. Song, Angew. Chem., Int. Ed., 2013, 52, 12143 CrossRef CAS; (p) S. Diosdado, J. Etxabe, J. Izquierdo, A. Landa, A. Mielgo, I. Olaizola, R. López and C. Palomo, Angew. Chem., Int. Ed., 2013, 52, 11846 CrossRef CAS.
- (a) Y. Sohtome, Y. Hashimoto and K. Nagasawa, Adv. Synth. Catal., 2005, 347, 1643 CrossRef CAS; (b) C. Uyeda and E. N. Jacobsen, J. Am. Chem. Soc., 2008, 130, 9228 CrossRef CAS; (c) M. P. Coles, Chem. Commun., 2009, 3659 RSC; (d) D. Leow and C. H. Tan, Synlett, 2010, 11, 1589 Search PubMed; (e) Y. Sohtome, N. Horitsugi, R. Takagi and K. Nagasawa, Adv. Synth. Catal., 2011, 353, 2631 CrossRef CAS; (f) X. Fu and C. H. Tan, Chem. Commun., 2011, 47, 8210 CAS; (g) M. Odagi, K. Furukori, T. Watanabe and K. Nagasawa, Chem.–Eur. J., 2013, 19, 16740 CrossRef CAS.
- (a) J. P. Malerich, K. Hagohara and V. H. Rawal, J. Am. Chem. Soc., 2008, 130, 14416 CrossRef CAS; (b) Y. Zhu, J. P. Malerich and V. H. Rawal, Angew. Chem., Int. Ed., 2010, 49, 153 CrossRef CAS; (c) Y. Qian, G. Ma, A. Lv, H. L. Zhu, J. Zhao and V. H. Rawal, Chem. Commun., 2010, 46, 3004 RSC; (d) D. Q. Xu, Y. F. Wang, W. Zhang, S. P. Luo, A. G. Zhong, A. B. Xia and Z. Y. Xu, Chem.–Eur. J., 2010, 16, 4177 CrossRef CAS; (e) L. Dai, S. Wang and F. Chen, Adv. Synth. Catal., 2010, 352, 2137 CrossRef CAS; (f) H. Y. Bae, S. Some, J. H. Lee, J. Kim, M. J. Song, S. Lee, Y. J. Zhang and C. E. Song, Adv. Synth. Catal., 2011, 353, 3196 CrossRef CAS; (g) W. Yang and D. Du, Adv. Synth. Catal., 2011, 353, 1241 CrossRef CAS; (h) J. Alemán, A. Parra, H. Jiang and K. A. Jøgensen, Chem.–Eur. J., 2011, 17, 6890 CrossRef; (i) R. I. Storer, C. Aciro and L. H. Jones, Chem. Soc. Rev., 2011, 40, 2330 RSC; (j) D. Mailhol, M. D. M. S. Duque, W. Raimondi, D. Bonne, T. Constantieux, Y. Coquerel and J. Rodriguez, Adv. Synth. Catal., 2012, 354, 3523 CrossRef CAS; (k) P. Kasaplar, P. Riente, C. Hartmann and M. A. Pericàs, Adv. Synth. Catal., 2012, 354, 2905 CrossRef CAS; (l) H. Yu, Q. Wang, Y. Wang, H. Song, Z. Zhou and C. Tang, Chem.–Asian J., 2013, 8, 2859 CrossRef CAS; (m) H. Wang, Y. Wang, H. Song, Z. Zhou and C. Tang, Eur. J. Org. Chem., 2013, 48441 Search PubMed; (n) T. Lu and S. E. Wheeler, Chem.–Eur. J., 2013, 19, 15141 CrossRef CAS; (o) Y. Liu, Y. Wang, H. Song, Z. Zhou and C. Tang, Adv. Synth. Catal., 2013, 355, 2544 CrossRef CAS.
- (a) K. P. Nair, V. Breedveld and M. Weck, Macromolecules, 2008, 41, 3429 CrossRef CAS; (b) F. Herbst, K. Schroeter, I. Gunkel, S. Groeger, T. Thurn-Albrecht, J. Balbach and W. H. Binder, Macromolecules, 2010, 43, 10006 CrossRef CAS; (c) K. P. Nair, V. Breedveld and M. Weck, Soft Matter, 2011, 7, 533 RSC; (d) S. Seiffert and J. Sprakel, Chem. Soc. Rev., 2012, 41, 909 RSC.
- (a) Y. Ducharme and J. D. Wuest, J. Org. Chem., 1988, 53, 5787 CrossRef CAS; (b) W. L. Jørgensen and J. Pranata, J. Am. Chem. Soc., 1990, 112, 2008 CrossRef; (c) J. Pranta, S. G. Wierschke and W. L. Jørgensen, J. Am. Chem. Soc., 1991, 113, 2810 CrossRef; (d) J. Sartorius and H. J. Schneider, Chem.–Eur. J., 1996, 2, 1446 CrossRef CAS; (e) F. H. Beijer, H. Kooijman, A. L. Spek, R. P. Sijbesma and E. W. Meijer, Angew. Chem., Int. Ed., 1998, 37, 75 CrossRef CAS.
- (a) W. K. Chan, W. Y. Yu, C. M. Che and M. K. Wong, J. Org. Chem., 2003, 68, 6576 CrossRef CAS; (b) Y. S. Fung, S. C. Yan and M. K. Wong, Org. Biomol. Chem., 2012, 10, 3122 RSC.
- G. L. Li, K. K. Y. Kung, L. Zou, H. C. Chong, Y. C. Leung, K. H. Wong and M. K. Wong, Chem. Commun., 2012, 48, 3527 RSC.
- (a) D. G. Barceloux, Barbiturates: Amobarbital, Butalbital, Pentobarbital, Secobarbital, in Medical Toxicology of Drug Abuse: Synthesized Chemicals and Psychoactive Plants, John Wiley and Sons, 2012, pp. 467–485 Search PubMed; (b) R. Michelucci, E. Pansini, C. Tassinari, Phenobarbital, Primidone and Other Barbiturates, in The Treatment of Epilepsy, Wiley-Blackwell, 3rd edn, 2009, pp. 585–603 Search PubMed; (c) N. L. Harrison and M. A. Simmonds, Br. J. Pharmacol., 1983, 80, 387 CrossRef CAS; (d) W. Löscher and M. A. Rogawski, Epilepsia, 2012, 53, 12 CrossRef; (e) M. Bialer, Epilepsia, 2012, 53, 3 CrossRef CAS.
- (a) K. Tanaka, X. Cheng, T. Kimura and F. Yoneda, Chem. Pharm. Bull., 1986, 34, 3945 CrossRef CAS; (b) K. Tanaka, X. Cheng and F. Yoneda, Tetrahedron, 1988, 44, 3241 CrossRef CAS; (c) A. Ikeda, Y. Kawabe, T. Sakai and K. Kaeasaki, Chem. Lett., 1989, 1803 CrossRef; (d) J. D. Figueroa-Villar, C. E. Rangel and L. N. Dos Santos, Synth. Commun., 1992, 22, 1159 CrossRef CAS.
- (a) M. Bauer and S. Spange, Angew. Chem., Int. Ed., 2011, 50, 9727 CrossRef CAS; (b) A. Niemz and V. M. Rotello, Acc. Chem. Res., 1999, 32, 44 CrossRef CAS; (c) S. S. Agasti, S. T. Caldwell, G. Cooke, B. J. Jordan, A. Kennedy, N. Kryvokhyzha, G. Rabani, S. Rana, A. Sanyalc and V. M. Rotello, Chem. Commun., 2008, 4123 RSC.
- I. Bolz, D. Schaarschmidt, T. Ruffer, H. Lang and S. Spange, Angew. Chem., Int. Ed., 2009, 48, 7440 CrossRef CAS.
- D. Benito-Garagorri, E. Becker, J. Wiedermann, W. Lackner, M. Pollak, K. Mereiter, J. Kisala and K. Kirchner, Organometallics, 2006, 25, 1900 CrossRef CAS.
- Z. Xu, P. Daka, I. Budik, H. Wang, F. Q. Bai and H. X. Zhang, Eur. J. Org. Chem., 2009, 4581 CrossRef CAS.
- M. Habel, C. Niederalt, S. Grimme, M. Nieger and F. Vogtle, Eur. J. Org. Chem., 1998, 1471 CrossRef CAS.
- J. Barluenga, J. M. Álvarez-Gutiérrez, A. Ballesteros and J. M. González, Angew. Chem., Int. Ed., 2007, 46, 1281 CrossRef CAS.
- (a) Z. Tian, A. Fattahi, L. Lis and S. R. Kass, J. Am. Chem. Soc., 2009, 131, 16984 CrossRef CAS; (b) A. Shokri, X. B. Wang and S. R. Kass, J. Am. Chem. Soc., 2013, 135, 9525 CrossRef CAS.
- K. M. Lippert, K. Hof, D. Gerbig, D. Ley, H. Hausmann, S. Guenther and P. R. Schreiner, Eur. J. Org. Chem., 2012, 5919 CrossRef CAS.
- All reactions were conducted with 0.025 mmol of benzylidene barbiturate 5a, 0.25 mmol of 2-methylfuran, 0.03 mmol of internal standard dichloromethane and 20 mol% of HB-DAD organocatalysts. The solutions were mixed thoroughly. The first NMR spectrum was taken 5 min after the addition of 2-methylfuran. Additional NMR spectra were recorded every 5 min for a total of 120 min.
- (a) A. Wittkopp and P. R. Schreiner, Chem.–Eur. J., 2003, 9, 407 CrossRef CAS; (b) P. N. H. Huynh, R. R. Walvoord and M. C. Kozlowski, J. Am. Chem. Soc., 2012, 134, 15621 CrossRef CAS.
- The calculation of relative rate constant of 1a, kobs(1a) = 0.000148 s−1, kobs(background) = kuncata = 0.0000377 s−1, kcat = kobs(1a) − kuncata = 0.000148 − 0.0000377 = 0.000111 s−1, krel = kcat/kuncata = 0.000111/0.000038 = 2.9 at 20 mol% of 1a.
- (a) K. A. Connors, Binding Constants: The Measurement of Molecular Complex Stability, Wiley-VCH, Weinheim, 1987 Search PubMed; (b) C. W. Davies, Ion association, Butterworths, Washington, 1962, ch. 4 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 991734. See DOI: 10.1039/c4ra04020a |
| This journal is © The Royal Society of Chemistry 2014 |