
Synthesis of biologically active pyridoimidazole/imidazobenzothiazole annulated polyheterocycles using cyanuric chloride in water†
Abstract
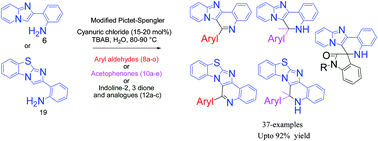
An efficient and mild protocol for rapid access to N-fused polyheterocycles via Pictet–Spengler type 6-endo cyclization using cyanuric chloride in an aqueous reaction medium has been developed. The protocol was successfully applied to a wide range of compounds including aryl/heteroaryl aldehydes (8a–o), ketones (10a–e), an electron-rich metallocene aldehyde (8e) and indoline-2,3-diones (12a–c) using cyanuric chloride (15–20 mol%) with tetra-n-butylammonium bromide (TBAB) (2.0 eq.) as an additive at 80–90 °C to give a good to excellent yield (66–92%) of polyheterocycles. Some of the synthesized compounds were found to exhibit antiplasmodial activity against chloroquine-sensitive (CQ-S) 3D7 and chloroquine-resistant (CQ-R) K1 strains of Plasmodium falciparum.
 Please wait while we load your content...
Please wait while we load your content...

