N-heterocyclic carbene-catalyzed oxidation of aldehydes for the synthesis of amides via phenolic esters†
Miran Jia,
Seungyeon Lima and
Hye-Young Jang*ab
aDivision of Energy Systems Research, Ajou University, Suwon 443-749, Korea. E-mail: hyjang2@ajou.ac.kr; Fax: +82 31 219 1615; Tel: +82 31 219 2555
bKorea Carbon Capture & Sequestration R&D Center, Deajeon 305-343, Korea
First published on 17th June 2014
Abstract
N-heterocyclic carbene-catalyzed oxidation using TEMPO is reported for the conversion of aldehydes to amides. A wide range of amides were synthesized in good yields (up to 72%) via a one-pot, sequential protocol involving oxidative esterification of aldehydes and subsequent aminolysis. To promote efficient aminolysis, various alkoxide leaving groups were evaluated.
The amide group is a common feature in many important compounds including pharmaceuticals, and in peptide bonds, and polymers (e.g., nylon and aramid).1 As a result, a variety of synthetic methods have been developed over the years to form amides, including use of coupling reagents, metal-catalysts, and metal-free conditions.2–7 Amongst these, our attention has been drawn to carbene-catalyzed amidation conditions because of the environmental benefits of metal-free conditions. Compared to carbene-catalyzed esterification of aldehydes,8–10 carbene-catalyzed amide formation from aldehydes is limited because of the competing imine formation. Based on Studer's report on carbene-catalyzed oxidative amidation, imine formation can be reduced using a sequential approach of carbene-catalyzed reactive ester formation, followed by aminolysis of the esters.6a,11 Although most carbene-catalyzed reactions required higher catalyst loadings and showed lower turnovers compared to those of the metal-catalyzed reactions, continuous studies of carbene-catalyzed reactions would provide efficient metal-free synthetic protocols.
We recently reported a carbene-catalyzed oxidation of aldehydes using 2,2,6,6-tetramethylpiperidinyloxy (TEMPO) to afford a diverse range of esters and thioesters.12 Under Studer's conditions, the stable TEMPO-ester formed readily, thereby making formation of the other esters and thioesters impossible.10a However, we found that we could modulate the formation of various esters and thioesters without forming the TEMPO-esters as the product. Herein, we report the use of carbene catalysts and TEMPO oxidant for the tandem oxidative esterification of aldehydes-aminolysis, to afford a diverse range of amides from aldehydes. A possible mechanism is proposed in Scheme 1. Based on our previous work, aldehyde 1a undergoes oxidative esterification via Breslow intermediate I to afford phenolic ester 1c. Subsequent aminolysis of 1c provides desired amide 1b.
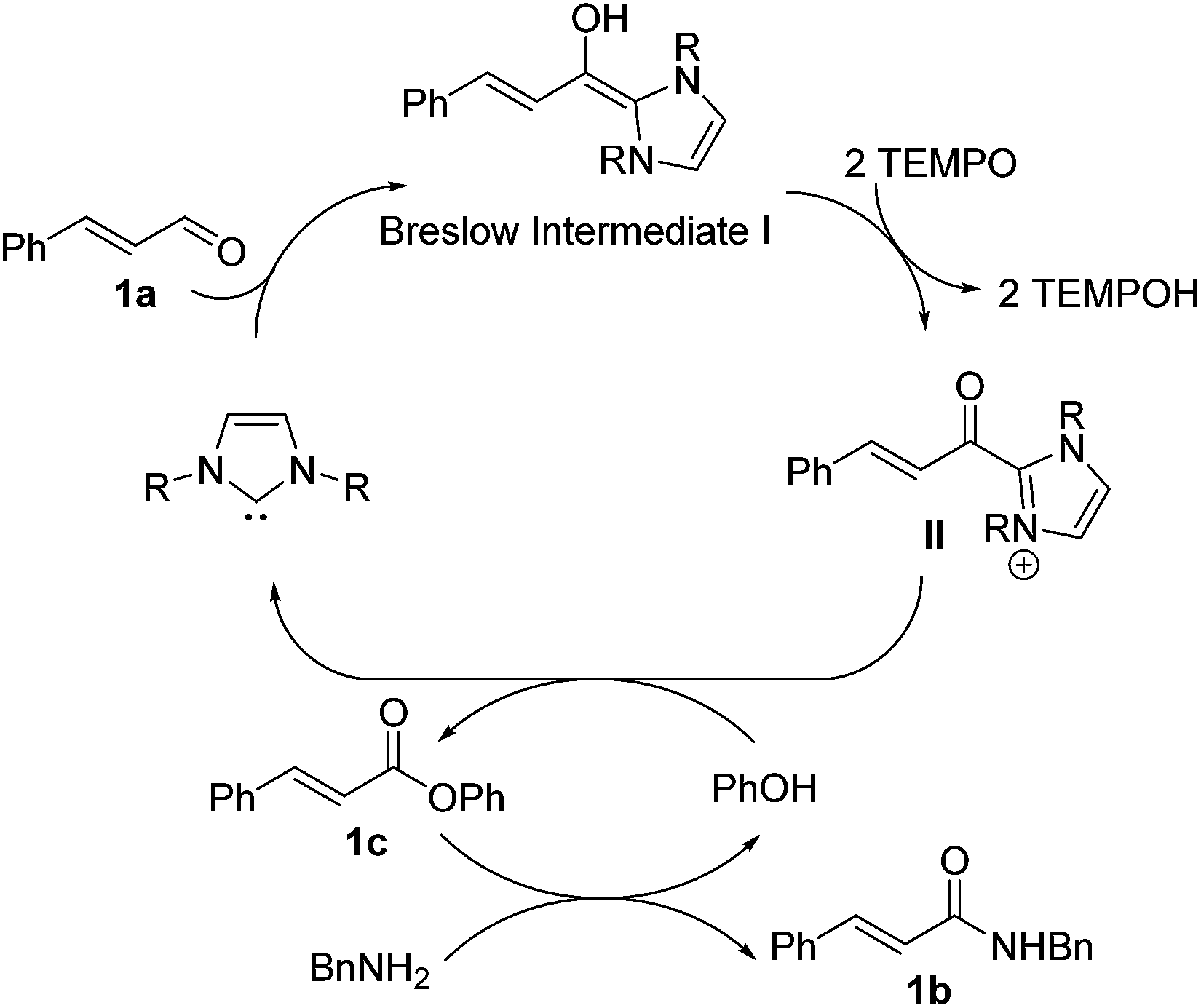 | ||
| Scheme 1 N-heterocyclic carbene-catalyzed amides synthesis from aldehydes. | ||
Optimization results are listed in Table 1. Initially, the oxidative esterification of cinnamaldehyde 1a with various alcohols, followed by aminolysis using benzyl amine was investigated. Alcohols (1 equiv.) were reacted with cinnamaldehyde 1a (1 equiv.) in the presence of 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene (IPr, 10 mol%), TEMPO (2 equiv.) in toluene at 100 °C for 4 h, followed by addition of benzyl amine (2 equiv.) and the mixture being stirred at 40 °C for 18 h. As shown in entry 1 of Table 1, reaction of 1a with isopropyl alcohol failed to form the desired amide 1b. Use of fluorinated alcohols improved the yield to 34% and 57% (entries 2 and 3). It was proposed that the basicity of the conjugated base of the alcohol could affect the progress of the aminolysis step, as the oxidative esterification of 1a to form the corresponding esters was completed within 4 h. Thus, the more basic isopropoxide group is considered as a poorer leaving group, and therefore no amide formation was observed. By considering the pKa of the alcohols, phenol, with a similar pKa to that of hexafluoroisopropyl alcohol (HFIP) was tested (entry 4).13 As expected, these reactions, which proceeded via the phenolic and hexafluoroisopropyl ester respectively, afforded the similar yields of product (entries 3 and 4). In addition to phenol, pNO2–phenol and pentafluorophenol (F5-PhOH) were tested; however, these failed to form desired amide 1b (entries 5 and 6). In the case of the electron-deficient phenols, the intermediate esters were not formed because of the low nucleophilicity of the phenol derivatives. It was found that the amount of TEMPO could be reduced to 1.5 equiv. and afforded amide 1b in 44% yield (entry 7). The IPr carbene catalyst loading was also reduced to 5 mol%; however, this led to a decrease in the yield of 1b (entry 8). Finally, an alternative carbene catalyst (IMes) was investigated but unfortunately this also led to a lower yield of 1b compared to that with the IPr catalyst (43%, entry 9).14 In the absence of carbene catalysts, either phenolic ester 1c or amide 1b was not formed. Without TEMPO, saturated amide 1d was formed in 69% yield, implying redox-esterification instead of oxidative esterification occurred in the absence of TEMPO.15 Based on our previous reports regarding the oxidative esterification of 1a with phenol,12 it was expected that excessive amounts of phenol would not increase the yield of esters. This was confirmed by reacting 1a with 1 and 2 equiv. of phenol which afforded 1c in a comparable 75% and 63% yields, respectively (Scheme 2). Subsequently, the stoichiometry of phenol was fixed at 1 equiv. with respect to 1a.
|
|||||
|---|---|---|---|---|---|
| Entry | Carbene | TEMPO | ROH | pKa of ROH | Yielda (1b) |
a Isolated yield.b Experimental: TEMPO (1.0 mmol) and IPr (0.05 mmol) was added to a solution of 1a (0.5 mmol) and ROH (0.5 mmol) in toluene (0.5 M) under nitrogen atmosphere. The reaction mixture was stirred at 100 °C for 4 h. Then, benzylamine (1.0 mmol) was added to the reaction vessel and the reaction mixture was stirred at 40 °C for 18 h. |
|||||
| 1 | IPr (10 mol%) | 2 equiv. | iPrOH | 17 | 0% |
| 2 | IPr (10 mol%) | 2 equiv. | CF3CH2OH | 12.4 | 34% |
| 3 | IPr (10 mol%) | 2 equiv. | HFIP | 9.3 | 57% |
| 4 | IPr (10 mol%) | 2 equiv. | PhOH | 10.0 | 60% |
| 5 | IPr (10 mol%) | 2 equiv. | pNO2–PhOH | 7.2 | 0% |
| 6 | IPr (10 mol%) | 2 equiv. | F5-PhOH | 5.5 | 0% |
| 7 | IPr (10 mol%) | 1.5 equiv. | PhOH | 10.0 | 44% |
| 8 | IPr (5 mol%) | 2 equiv. | PhOH | 10.0 | 44% |
| 9 | IMes (10 mol%) | 2 equiv. | PhOH | 10.0 | 43% |
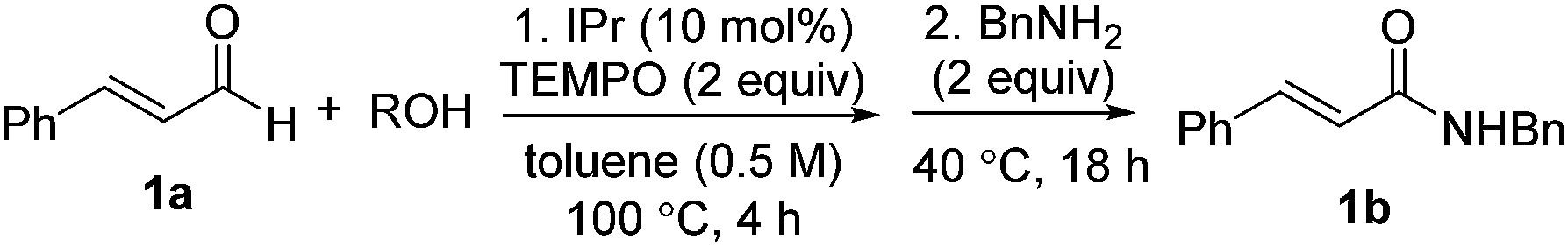
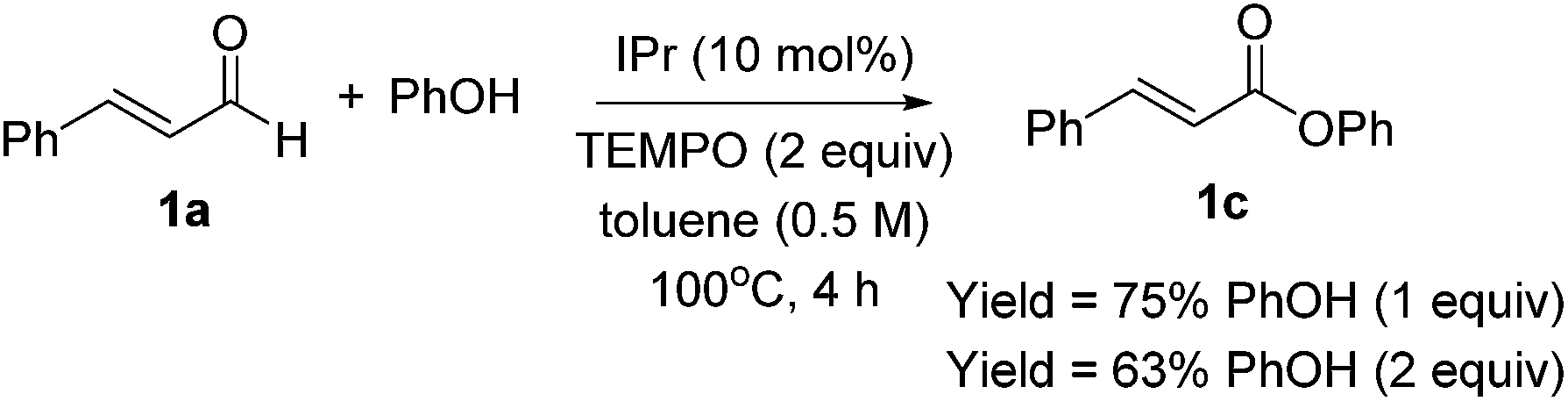 | ||
| Scheme 2 Oxidative esterification of 1a with phenol. | ||
Next, the substrate scope was investigated by employing a diverse range of amines and aldehydes (Tables 2 and 3). The reaction of cinnamaldehyde with various amines was conducted using the optimized reaction conditions. Electron-rich benzyl amines (p-methyl benzyl amine and p-methoxy benzyl amine) performed well in the reaction with cinnamaldehyde, to afford 2b and 3b in 55% and 59% yield, respectively (Table 2, entries 1 and 2). Heteroaromatic amine led to the formation of 4b in a slightly reduce yield (41%, entry 3). The reactions of cinnamaldehyde with allyl amine and aliphatic amine proceeded well to afford 5b (50%) and 6b (52%) (entries 4 and 5). An amide formation using sterically hindered α-ethylbenzyl amine was also successful and afforded 7b in 60% yield (entry 6).
|
|||
|---|---|---|---|
| Entry | Amine | Product | Yielda |
| a Isolated yield.b Experimental: TEMPO (1.0 mmol) and IPr (0.05 mmol) was added to a solution of 1a (0.5 mmol) and PhOH (0.5 mmol) in toluene (0.5 M) under nitrogen atmosphere. The reaction mixture was stirred at 100 °C for 4 h. Then, benzylamine (1.0 mmol) was added to the reaction vessel and the reaction mixture was stirred at 40 °C for 18 h. | |||
| 1 | 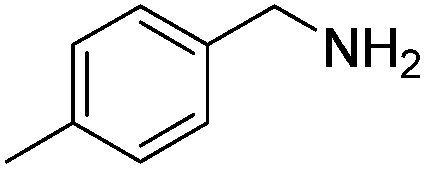 |
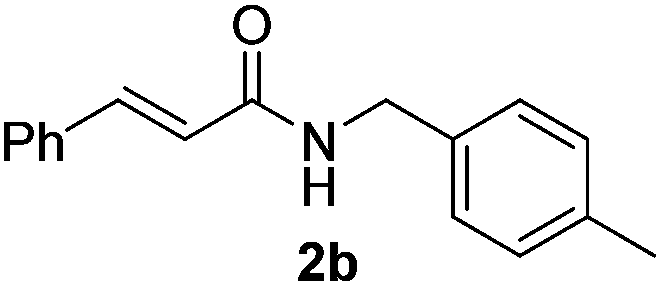 |
55% |
| 2 | 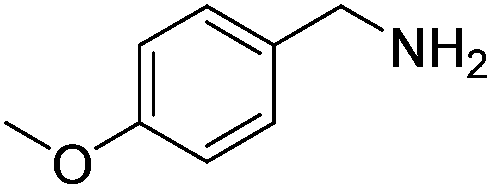 |
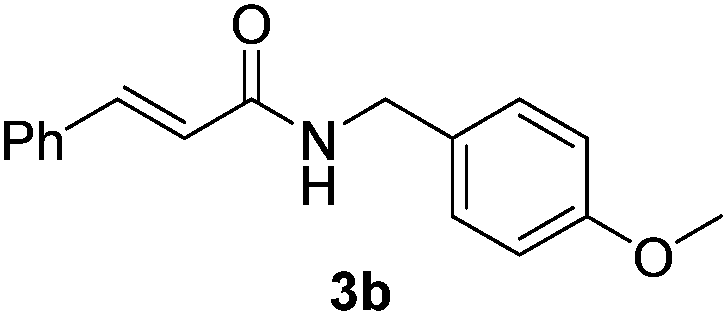 |
59% |
| 3 | 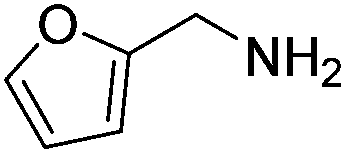 |
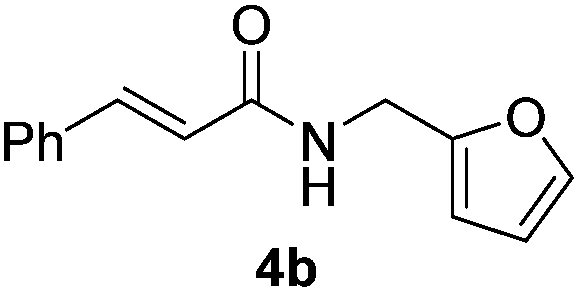 |
41% |
| 4 | 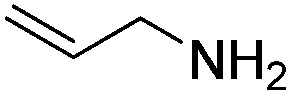 |
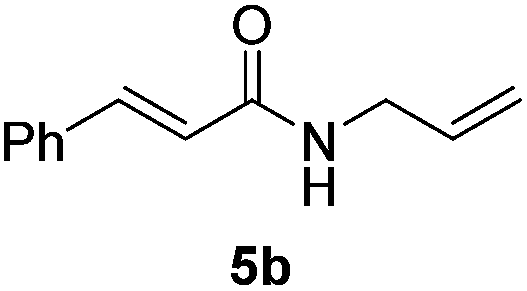 |
50% |
| 5 | 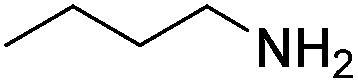 |
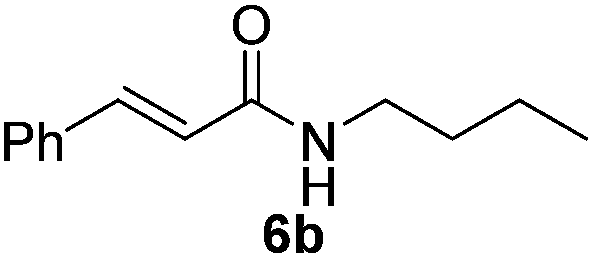 |
52% |
| 6 | 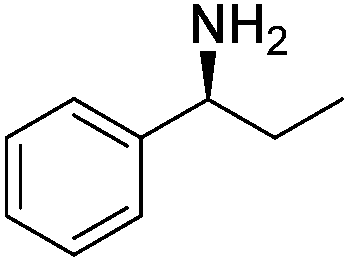 |
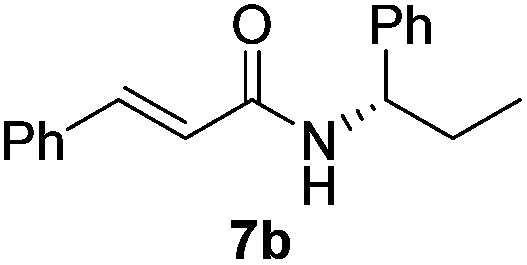 |
60% |
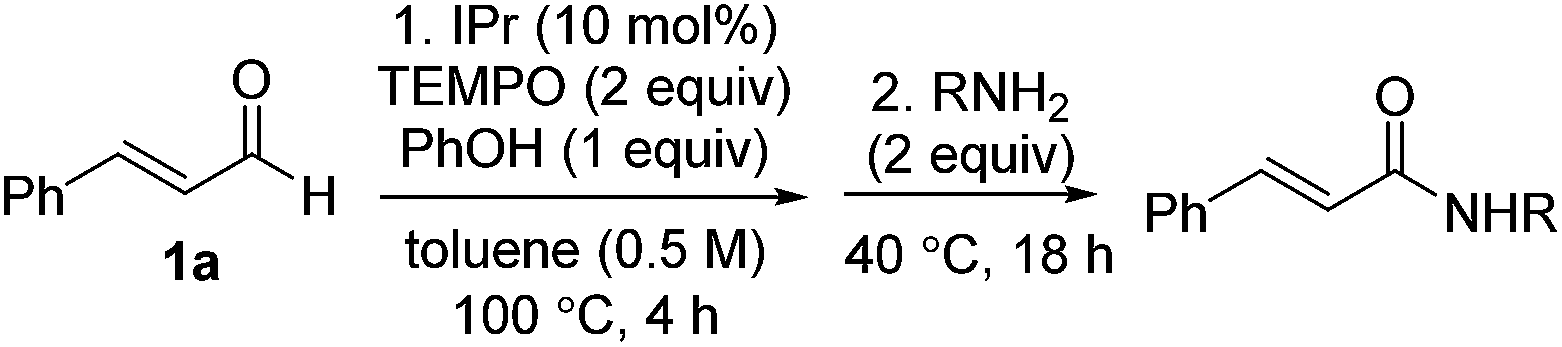
|
|||
|---|---|---|---|
| Entry | Amine | Product | Yielda |
| a Isolated yield.b Experimental: TEMPO (1.0 mmol) and IPr (0.05 mmol) was added to a solution of aldehyde (0.5 mmol) and PhOH (0.5 mmol) in toluene (0.5 M) under nitrogen atmosphere. The reaction mixture was stirred at 100 °C for 4 h. Then, benzylamine (1.0 mmol) was added to the reaction vessel and the reaction mixture was stirred at 40 °C for 18 h. | |||
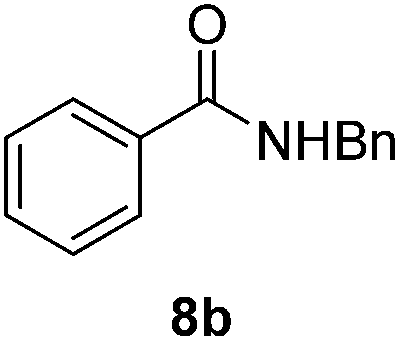
| 1 |  |
 |
55% |
| 2 |  |
 |
55% |
| 3 | 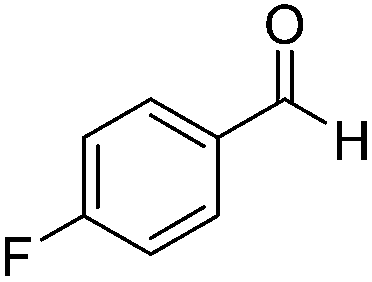 |
 |
56% |
| 4 | 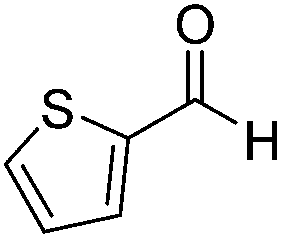 |
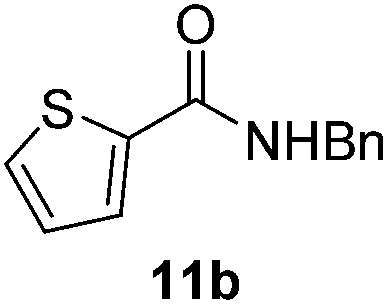 |
54% |
| 5 | 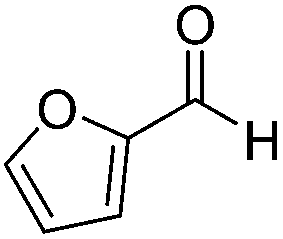 |
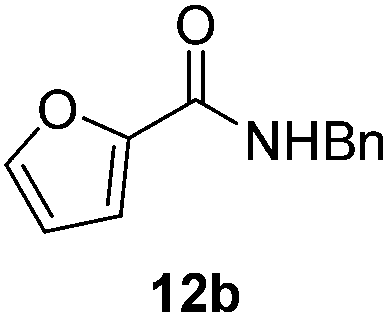 |
72% |
| 6 | 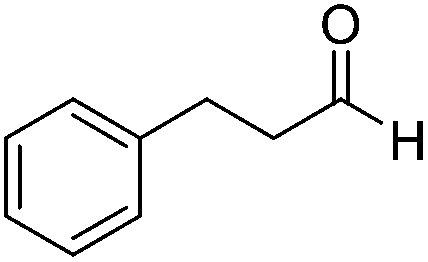 |
 |
45% |
| 7 | 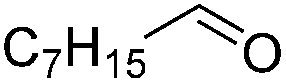 |
 |
38% |
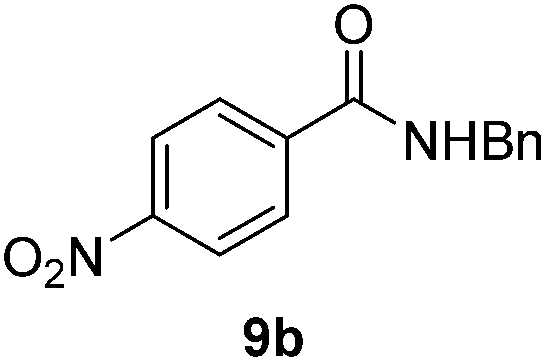
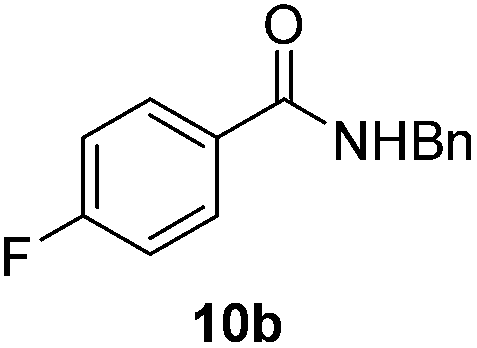
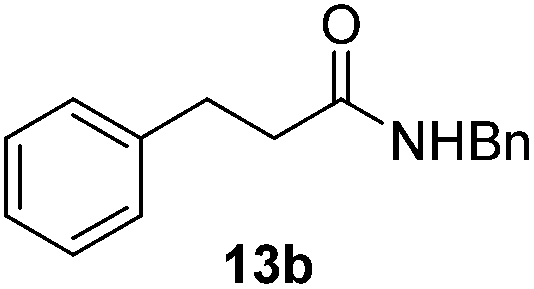
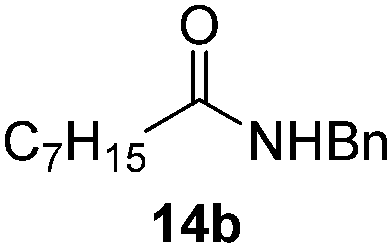
Next, the scope of aldehyde was investigated for amide formation using benzyl amine (Table 3). Benzaldehyde and electron-deficient benzaldehydes (pNO2-substituted and pfluoro-substituted) reacted with benzyl amine to provide 8b (55%), 9b (55%), and 10b (56%), respectively (entries 1–3). Thiophenyl carboxaldehyde and furfural reacted well to afford 11b and 12b in 54% and 72% yield, respectively (entries 4 and 5). In addition to aromatic aldehydes, aliphatic aldehydes were also subjected to the reaction conditions; however, this afforded aliphatic amide 13b and 14b in a reduced 45% and 38% yield, respectively (entries 6 and 7).
Conclusions
In conclusion, we have expanded our NHC-catalyzed oxidative coupling using TEMPO for the synthesis of a range of amides from aldehydes. To address the previous imine formation issues, we utilized a tandem reaction protocol involving NHC-catalyzed oxidative phenolic ester formation followed by aminolysis. The optimum alcohol for the ester formation and aminolysis was chosen based on pKa values. As a result, phenol (pKa = 10.0) was found to be the most favourable alcohol for the amide formation. Under optimized conditions, a diverse range of aromatic and aliphatic aldehydes and amines were coupled to form the desired amides in modest to good yield via the intermediate esters.Acknowledgements
This study was supported by the Korea Research Foundation (no. 2009-0094046 and 2013008819) and the Korea CCS R&D Center (KCRC) grant funded by the Korea Government (Ministry of Education, Science and Technology) (no. 2012-0008935).Notes and references
- (a) A. K. Ghose, V. N. Viswanadhan and J. J. Wendoloski, J. Comb. Chem., 1999, 1, 55–68 CrossRef CAS; (b) J. W. Bode, Curr. Opin. Drug Discovery Dev., 2006, 9, 765–775 CAS; (c) T. Cupido, J. Tulla-Puche, J. Spengler and F. albericio, Curr. Opin. Drug Discovery Dev., 2007, 10, 768–783 CAS.
- For selected review articles for the synthesis of amides, see: (a) S.-Y. Han and Y.-A. Kim, Tetrahedron, 2004, 60, 2447–2467 CrossRef CAS; (b) C. A. G. N. Montalbetti and V. Falque, Tetrahedron, 2005, 61, 10827–10852 CrossRef CAS; (c) K. Ekoue-Kovi and C. Wolf, Chem.–Eur. J., 2008, 14, 6302–6315 CrossRef CAS PubMed; (d) A. El-Faham and F. Albericio, Chem. Rev., 2011, 111, 6557–6602 CrossRef CAS PubMed; (e) V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471–479 CrossRef CAS PubMed; (f) S. Roy, S. Roy and G. W. Gribble, Tetrahedron, 2012, 68, 9867–9923 CrossRef CAS.
- For selected articles for the transition metal-catalyzed amidation of alcohols, see: (a) C. Gunanathan, Y. Ben-David and D. Milstein, Science, 2007, 317, 790–792 CrossRef CAS; (b) L. U. Nordstrøm, H. Vogt and R. Madsen, J. Am. Chem. Soc., 2008, 130, 17672–17673 CrossRef PubMed; (c) S. C. Ghosh, S. Muthaiah, Y. Zhang, X. Xu and S. H. Hong, Adv. Synth. Catal., 2009, 351, 2643–2649 CrossRef CAS; (d) S. Muthaiah, S. C. Ghosh, J.-E. Jee, C. Chen, J. Zhang and S. H. Hong, J. Org. Chem., 2010, 75, 3002–3006 CrossRef CAS PubMed; (e) Y. Wang, D. Zhu, L. Tang, S. Wang and Z. Wang, Angew. Chem., Int. Ed., 2011, 50, 8917–8921 CrossRef CAS PubMed; (f) J.-F. Soule, H. Miyamura and S. Kovayashi, J. Am. Chem. Soc., 2011, 133, 18550–18553 CrossRef CAS PubMed; (g) S. Kegnæs, J. Mielby, U. V. Mentzel, T. Jensen, P. Fristrup and A. Riisager, Chem. Commun., 2012, 48, 2427–2429 RSC; (h) S. C. Ghosh, J. S. Y. Ngiam, A. M. Seayad, D. T. Tuan, C. W. Johannes and A. Chen, Tetrahedron Lett., 2013, 54, 4922–5492 CrossRef CAS.
- For selected articles for the transition metal-catalyzed oxidative amidation of aldehydes, see: (a) A. Tillack, I. Rudloff and M. Beller, Eur. J. Org. Chem., 2001, 523–528 CrossRef CAS; (b) W.-J. Yoo and C.-J. Li, J. Am. Chem. Soc., 2006, 128, 13064–13065 CrossRef CAS PubMed; (c) J. W. W. Chang and P. W. H. Chan, Angew. Chem., Int. Ed., 2008, 47, 1138–1140 CrossRef CAS; (d) S. Seo and T. J. Marks, Org. Lett., 2008, 10, 317–319 CrossRef CAS PubMed; (e) L. Wang, H. Fu, Y. Jiang and Y. Zhao, Chem.–Eur. J., 2008, 14, 10722–10726 CrossRef CAS PubMed; (f) C. Qian, X. Zhang, J. Li, F. Xu, Y. Zhang and Q. Shen, Organometallics, 2009, 28, 3856–3862 CrossRef CAS; (g) J. Li, F. Xu, Y. Zhang and Q. Shen, J. Org. Chem., 2009, 74, 2575–2577 CrossRef CAS PubMed; (h) S. C. Ghosh, J. S. Y. Ngiam, C. L. L. Chai, A. M. Seayad, T. T. Dang and A. Chen, Adv. Synth. Catal., 2012, 354, 1407–1412 CrossRef CAS; (i) S. C. Ghosh, J. S. Y. Ngiam, A. M. Seayad, D. T. Tuan, C. L. L. Chai and A. Chen, J. Org. Chem., 2012, 77, 8007–8015 CrossRef CAS PubMed; (j) C. Zhang, X. Zong, L. Zhang and N. Jiao, Org. Lett., 2012, 14, 3280–3283 CrossRef CAS PubMed; (k) H. Yao and K. Yamamoto, Chem.–Asian J., 2012, 7, 1542–1545 CrossRef CAS PubMed.
- For the oxidative amidation of aldehydes in the absence of carbene catalysts, see: (a) K. Ekoue-Kovi and C. Wolf, Org. Lett., 2007, 9, 3429–3432 CrossRef CAS PubMed; (b) J.-J. Shie and J.-M. Fang, J. Org. Chem., 2003, 68, 1158–1160 CrossRef CAS PubMed; (c) K. Xu, Y. Hu, S. Zhang, Z. Zhan and Z. Wang, Chem. Eur. J., 2012, 18, 9793–9797 CrossRef CAS PubMed.
- For carbene-catalyzed oxidative amidation of aldehydes, see: (a) S. D. Sarkar and A. Studer, Org. Lett., 2010, 12, 1992–1995 CrossRef PubMed; (b) S. Kuwano, S. Harada, R. Oriez and K.-i. Yamada, Chem. Commun., 2012, 48, 145–147 RSC.
- For carbene-catalyzed redox amidation of aldehydes, see: (a) H. U. Vora and T. Rovis, J. Am. Chem. Soc., 2007, 129, 13796–13797 CrossRef CAS PubMed; (b) J. W. Bode and S. S. Sohn, J. Am. Chem. Soc., 2007, 129, 13798–13799 CrossRef CAS PubMed; (c) H. U. Vora, P. Wheeler and T. Rovis, Adv. Synth. Catal., 2012, 354, 1617–1639 CrossRef CAS PubMed.
- For reviews of NHC-catalyzed reactions, see: (a) D. Enders and T. Balensiefer, Acc. Chem. Res., 2004, 37, 534–541 CrossRef CAS PubMed; (b) K. Zeitler, Angew. Chem., Int. Ed., 2005, 44, 7506–7510 CrossRef CAS PubMed; (c) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606–5655 CrossRef CAS PubMed; (d) N. Marion, S. Díez-González and S. P. Nolan, Angew. Chem., Int. Ed., 2007, 46, 2988–3000 CrossRef CAS PubMed; (e) H. U. Vora and T. Rovis, Aldrichimica Acta, 2011, 44, 3 CAS; (f) J. Mahatthananchai and J. W. Bode, Chem. Sci., 2012, 3, 192–197 RSC; (g) S. De Sarkar, A. Biswas, R. C. Samanta and A. Studer, Chem.–Eur. J., 2013, 19, 4664–4678 CrossRef CAS PubMed , Carbene-catalyzed oxidation.
- For carbene-catalyzed oxidation of aldehydes to carboxylic acids, see: P.-C. Chiang and J. W. Bode, Org. Lett., 2011, 13, 2422–2425 CrossRef CAS PubMed.
- For carbene-catalyzed oxidative esterification of aldehydes, see: (a) J. Guin, S. De sarkar, S. Grimme and A. Studer, Angew. Chem., Int. Ed., 2008, 47, 8727–8730 CrossRef CAS PubMed; (b) B. E. Maki and K. A. Scheidt, Org. Lett., 2008, 10, 4331–4334 CrossRef CAS; (c) C. Noonan, L. Baragwanath and S. J. Cannon, Tetrahedron Lett., 2008, 49, 4003–4006 CrossRef CAS; (d) B. E. Maki, A. Chan, E. M. Phillips and K. A. Scheidt, Tetrahedron, 2009, 65, 3102–3109 CrossRef CAS PubMed; (e) S. De Sarkar, S. Grimme and A. Studer, J. Am. Chem. Soc., 2010, 132, 1190–1191 CrossRef CAS; (f) C. A. Rose and K. Zeitler, Org. Lett., 2010, 12, 4552–4555 CrossRef CAS PubMed; (g) B. Maji, S. Vedachalan, X. Ge, S. Cai and X.-W. Liu, J. Org. Chem., 2011, 76, 3016–3023 CrossRef CAS; (h) E. E. Finney, K. A. Ogawa and A. J. Boydston, J. Am. Chem. Soc., 2012, 134, 12374–12377 CrossRef CAS PubMed; (i) P. Arde, B. T. Ramanjaneyulu, V. Reddy, A. Saxena and R. V. Anand, Org. Biomol. Chem., 2012, 10, 848–851 RSC; (j) T. Uno, T. Inokuma and Y. Takemoto, Chem. Commun., 2012, 48, 1901–1903 RSC; (k) I. N. C. Kiran, K. Lalwani and A. Sudalai, RSC Adv., 2013, 3, 1695–1698 RSC; (l) L. Möhlmann, S. Ludwig and S. Blechert, Beilstein J. Org. Chem., 2013, 9, 602–607 CrossRef; (m) J. Zhao, C. Mück-Lichtenfeld and A. Studer, Adv. Synth. Catal., 2013, 355, 1098–1106 CrossRef CAS.
- For carbene-catalyzed amidation of unactivated esters, see: M. Movassaghi and M. A. Schmidt, Org. Lett., 2005, 7, 2453–2456 CrossRef CAS.
- M. Ji, X. Wang, Y. N. Lim, Y.-W. Kang and H.-Y. Jang, Eur. J. Org. Chem., 2013, 7881–7885 CrossRef CAS.
- For references presenting pKa values of alcohols, see: (a) W. A. Sheppard, J. Am. Chem. Soc., 1970, 92, 5419–5422 CrossRef CAS; (b) L. Eberson, M. P. Hartshorn, O. Persson and F. Radner, Chem. Commun., 1996, 2105–2112 RSC; (c) Y.-M. Shen, W.-L. Duan and M. Shi, Adv. Synth. Catal., 2003, 345, 337–340 CrossRef CAS.
- In our previous publication (ref. 12), the IPr carbene showed better activitity than the IMes carbene in the oxidative esterification of aldehydes. However, in the one-pot reaction involving oxidation of allylic alcohols and oxidative esterification, IMes showed better conversion than IPr. The catalytic activity of carbenes seems different depending on reaction conditions.
- In the absence of TEMPO, redox esterification occurred to afford saturated amide 1d
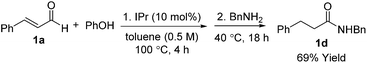 .
.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental and spectral analysis for new compounds. See DOI: 10.1039/c4ra04012k |
| This journal is © The Royal Society of Chemistry 2014 |