DOI:
10.1039/C4RA03908D
(Paper)
RSC Adv., 2014,
4, 33614-33618
A novel highly sensitive and selective fluorescent sensor for imaging mercury(II) in living cells†
Received
29th April 2014
, Accepted 28th July 2014
First published on 29th July 2014
Abstract
A novel phenothiazine-based derivative was designed and synthesized in this work, this molecule exhibited an obvious fluorescence quenching upon addition of Hg2+ due to the formation of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 metal–ligand complex. In addition to the high sensitivity toward Hg(II), it also displayed a high selectivity for Hg2+ over other transition metal ions in a rapid response time (<10 s). Simultaneously, the cell imaging experiment demonstrated the value of the sensor in fluorescent visualization of Hg2+ in biological systems.
:1 metal–ligand complex. In addition to the high sensitivity toward Hg(II), it also displayed a high selectivity for Hg2+ over other transition metal ions in a rapid response time (<10 s). Simultaneously, the cell imaging experiment demonstrated the value of the sensor in fluorescent visualization of Hg2+ in biological systems.
1. Introduction
Mercury is one of the most hazardous elements in ecosystems which is released through natural events or human activities.1 Mercury accumulation in the human body will affect a wide variety of diseases like digestive, kidney, and especially neurological diseases, even in a low concentration.2 Unfortunately, mercury contamination is widespread and occurs in the environment through a variety of natural and anthropogenic sources,3 and a high percentage of mercury contamination can be attributed to the industrial processes such as oceanic and volcanic emissions, coal-burning power plants, gold mining, solid waste incineration and the combustion of fossil fuels.4 Therefore, developing the real-time Hg2+ sensors with high sensitivity and selectivity are in great demand.
The development of fluorescent chemosensors for sensing and reporting heavy transition-metal ions has been receiving considerable attention in recent years.5 Until now, many fluorescent sensors for Hg2+ have been reported, most of them are based on common fluorophore such as rhodamine or naphthylamine derivatives,6 which are often hampered by interference from chemically related cations like Cu2+ and exhibit relatively long response times, and the phenothiazine based fluorescent sensor is rarely reported in early literatures.7 In these regards, our synthesis of the novel phenothiazine based fluorescent sensor which displays extremely selective and sensitive for Hg2+ is of great advantage. Besides, the rapid response time make it as a perfect tool for illustrating and dynamically mapping the intracellular fluctuations of Hg2+ by utilizing microscopy techniques to allow real-time local imaging.8
Herein we report a new fluorescent sensor combine quinoline with phenothiazine, which serving as a Hg2+ chelator based on ICT (intramolecular charge transfer) mechanism, presented very excellent affinity for Hg2+ among other metal ions and the living cells image experiments illustrated its favourable biocompatibility, in addition, its simple structure and rapid response will expand its application prospects.
2. Experimental
2.1 Materials and instruments
All the materials for synthesis were purchased from commercial suppliers and used without further purification. Solutions for spectra detection was HPLC reagent without fluorescent impurity. 1H NMR spectra were taken on a Varian mercury-400 spectrometer with TMS as an internal standard and DMSO as solvent. HRMS spectra was analysed on an Agilent 1290-micro TOF QII. Fluorescence spectra measurements were performed on a Hitachi F-4500 spectrofluorimeter. The pH measurements were made with Mettler-Toledo Instruments DELTE 320 pH. Cell experiment were applied on an inverted fluorescence microscope (Olympus IX-70) connected with a digital camera (Olympus, c-5050).
2.2 UV-vis and fluorometric analysis
The solution of sensor 1 was prepared in C2H5OH and the metal ion buffer solutions were using various cations dissolved in Tris–HCl buffer solution at pH 7.2. In titration and selectivity experiments, the test samples were prepared by placing appropriate amounts of ions into corresponding solution of sensor 1. For fluorescence measurements, excitation was provided at 368 nm, and emission was collected from 330 to 550 nm, both the excitation and emission slit widths were 5 and 5 nm, respectively.
2.3 Synthesis
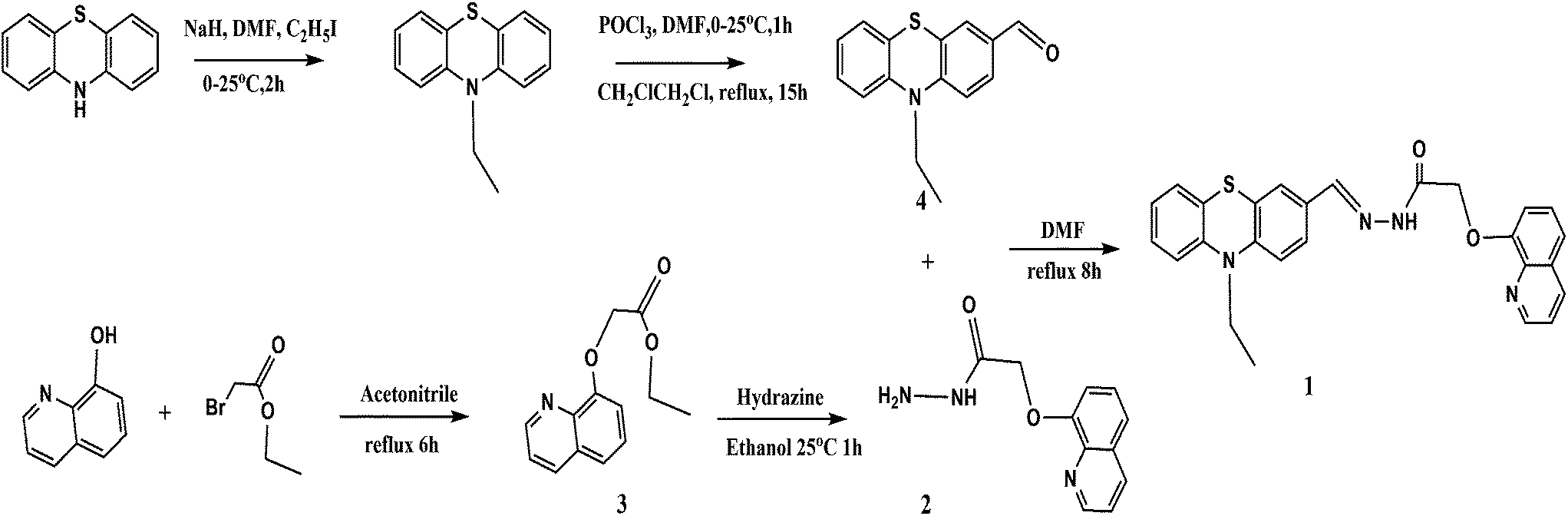
Preparation of intermediate 4. Phosphoryl chloride of 3.0 g (19 mmol) was added slowly to 2 mL of dry N,N-dimethylformamide (DMF) at 0 °C, and the mixture was stirred for 2 h at room temperature. Then, a solution of 10-ethylphenothiazine (2.4 g, 9.5 mmol) in 1,2-dichloroethane (25 mL) was added to it. After another 1 h, the reaction mixture was heated to 90 °C for 15 h. Finally, the solution was cooled and poured into 300 mL of water. After the solution was extracted with CH2Cl2 (70 mL × 3) and dried with anhydrous magnesium sulfate, the solvent was removed and the residue was purified by column chromatography with petroleum ether–ethyl acetate(15:1, volume ratio) as eluent to afford 1.75 g of compound 4 as a yellow solid, yield 32%.δ 1.31 (m, J = 6.0, 5.9 Hz, 3H), 3.96 (d, J = 3.1 Hz, 2H), 7.03 (m, J = 9.2, 5.7, 6.2 Hz, 1H), 7.15 (m, J = 6.5, 8.9 Hz, 1H), 7.55 (s, 1H), 7.70 (d, J = 6.3 Hz, 1H), 9.78 (s, 1H). 13C NMR (75 MHz DMSO, 25 °C, TMS): δ 12.91, 42.22, 115.49, 116.52, 122.26, 123.30, 123.98, 127.58, 128.01, 128.42, 130.70, 131.22, 143.02, 149.95, 190.94.
Preparation of intermediate 2 (ref. 9). (2.0 g, 13.78 mmol) 8-hydroxyquinoline and (3.8 g, 27.56 mmol) K2CO3 were mixed in 50 mL acetonitrile with stirring for 30 min, added (2.5 g, 15.15 mmol) ethyl bromoacetate and kept stirring for 6 h at room temperature, extracted the product with CH2Cl2 and H2O for 3 times, retained organic layer and dried on MgSO4 for 12 h before distilling the solvent. The crude product compound 3 (red oil) was purified by column chromatography (silica gel, EtOAc:petroleum ether = 1:2) with a yield of 80% [18]. (0.5 g, 2.16 mmol) compound 3 in 5 mL MeOH was added dropwise into 1 mL hydrazine for stirring 1 h, filtered the generated precipitates and washed with H2O and CH2Cl2 for 3 times respectively, purified the residue through silica gel column chromatography (silica gel, dichloromethane:ethanol = 1:1). The product intermediate 2 was collected as white solid with a yield of 69% (Scheme 1). 1H NMR (300 MHz DMSO, 25 °C, TMS): δ 4.39 (s, 2H), 4.76 (s, 2H), 7.26 (d, J = 6.0 Hz, 1H), 7.53 (d, J = 3.0 Hz, 2H), 7.59 (s, H), 8.36 (d, J = 3.9 Hz, 1H), 8.91 (d, J = 1.6 Hz, 1H), 9.46 (s, 1H). 13C NMR (75 MHz DMSO, 25 °C, TMS): δ 68.77, 112.19, 121.42, 122.48, 127.23, 129.6, 136.51, 140.33, 149.88, 154.42, 167.32. LC-MS. Calc. for: C11H11N3O2 m/z = 217.09, found [(M + 1)]+ m/z = 218.1.
 |
| | Scheme 1 Synthetic route of sensor 1. | |
Synthesis of sensor 1. To 20 mL of anhydrous DMF containing intermediate 4 (0.22 g, 1.0 mmol) was added intermediate 2 (0.25 g, 1.0 mmol) and the mixture was vigorously at 155 °C for 8 h. The reaction progress was monitored by thin-layer chromatography. After completion of the reaction, removed the solvent and the residue was purified by column chromatography (silica gel, dichloromethane:methanol = 2:1) to afford 0.11 g of sensor 1 as yellow solid, yield 23%. 1H NMR (300 M DMSO, 25 °C, TMS): δ 1.44 (m, J = 5.8, 5.9 Hz, 3H), 3.95 (d, J = 3.0 Hz, 2H), 4.99 (s, 2H), 6.89 (m, J = 6.2, 2.9, 3.2 Hz, 3H), 7.16 (m, J = 5.9, 8.8 Hz, 2H), 7.31 (m, J = 6.4, 2.7 Hz, 2H), 7.57 (m, J = 3.0, 3.3 Hz, 5H), 8.25 (m, J = 6.3, 5.7 Hz, 2H), 8.96 (d, J = 3.6 Hz, 1H). 13C NMR (75 MHz DMSO, 25 °C, TMS): δ 13.00, 41.83, 69.59, 110.38, 115.72, 115.78, 116.11, 120.37, 121.82, 123.29, 127.11, 127.32, 127.53, 128.24, 128.76, 136.23, 136.69, 143.36, 143.99, 146.09, 147.23, 148.39, 149.39, 149.88, 154.53, 169.18. LC-MS. Calc. for: C26H22N4O2S m/z = 454.15.13, found [(M + 1)+] m/z = 455.1.
3. Results and discussion
3.1 Hg2+-titration and spectral responses
With the synthesis complete, the optical property of sensor 1 in the presence of Hg2+ was investigated. Evidence for ion interaction with the chromophore was first sought using UV-visible spectroscopy, titration experiments were conducted under the condition of 0.01 mol L−1 Tris–HCl buffer solution at pH = 7.2. As illustrated in Fig. 1, sensor 1 exhibited an absorption maximum at 368 nm, with the addition of a Hg2+ buffer solution ([Hg2+] = 0–0.01 mmol L−1), the absorption band at 368 nm increased until the first stoichiometry. In the fluorescence emission (Fig. 2), sensor 1 (5 × 10−6 mol L−1) alone exhibited strong fluorescence at 526 nm (V(C2H5OH)–V(buffer) = 7:3, pH = 7.2, excited at 368 nm). Upon the addition of Hg2+ from 0 to 1 × 10−5 mol L−1, the fluorescence intensity gradually weakened until quenching and the reaction responsible for these changes reached completion well within the time frame (<10 s), this “switch-off” process could be observed under the irradiation of ultraviolet lamp (Scheme 2). The decreasing fluorescence intensity of sensor 1 depend on the concentration of Hg2+ was in a linear manner as illustrated in Fig. 3 (R = 0.99012), which indicated sensor 1 had potential application for quantitative determination of Hg2+, and the detecting limit could reach to 3.139 × 10−7 mol L−1 by calculation from this linear relationship (based on DL = KSb1/S). Simultaneously, this 10−7 mol L−1 degree detecting limit is much lower than the TLV (10 ppb) set by the EPA. The Job's plot (Fig. 4) exhibited the maximum absorbance appeared at w = 50%, demonstrated that sensor 1 and Hg2+ formed a 1:1 stoichiometry complex. Furthermore, the association constant of complex of sensor 1 with Hg2+ was found to be 8.667 × 106 mol−1 (R = 0.99611) according to the linear Benesi–Hildebrand (Fig. 5) expression [10–12], which based on the relationship between absorbance [1/(A − A0)] and 1/[Hg2+] at 368 nm in absorption spectrum.10
 |
| | Fig. 1 UV-Vis absorption response of sensor 1 (1 μM L−1) upon addition of different concentrations of Hg2+ (0.1 μM L−1) in Tris–HCl solution [V(C2H5OH)–V(H2O) = 7:3, pH = 7.2]. | |
 |
| | Fig. 2 Fluorescence spectra of sensor 1 (1 μM L−1) upon addition of different concentrations of Hg2+ (0.1 μM L−1) in Tris–HCl solution [V(C2H5OH)–V(H2O) = 7:3, pH = 7.2] (λex = 368 nm). | |
 |
| | Scheme 2 | |
 |
| | Fig. 3 Normalized response of the fluorescent signal to changing Hg2+ concentrations. | |
 |
| | Fig. 4 Job's plot for determining the stoichiometry of sensor 1 and Hg2+ in Tris–HCl solution [V(C2H5OH)–V(H2O) = 7:3, pH = 7.2] [Hg2+] + [sensor 1], the total concentration of sensor 1 and Hg2+ was 1 × 10−5 mol L−1. | |
 |
| | Fig. 5 Benesi–Hildebrand plot of sensor 1 with Hg2+. | |
To gain the insight into the observed fluorescence quenching, we may consider that the specific binding between sensor 1 and Hg2+ change the stereochemical structure of sensor 1, and then interrupted the π-conjugation and affected the intramolecular electron density distribution, resulting in the fluorescence quenching to Hg2+ in aqueous solution.11 The observation was in good agreement with the afore-mentioned design concept.
3.2 Selective and competitive experiments
To gain insight into the selectivity of sensor 1 for Hg2+, various common metal ions in environmental and biological interest were introduced to investigate their impact on the fluorescence response of sensor 1. In selectivity experiments the fluorometric behavior of sensor 1 (5.0 × 10−6 mol L−1) was investigated upon addition of several metal ions such as Na+, K+, Ca2+, Mg2+, Cu2+, Ba2+, Fe3+, Mn2+, Fe2+, Pb2+, Cd2+, Hg2+, Zn2+, Al3+ (5.0 × 10−6 mol L−1) in C2H5OH–Tris buffer solution [V(C2H5OH)–V(H2O) = 7:3, pH 7.2, excited at 368 nm]. As shown in Fig. 6 (black bar), only Hg2+ to sensor 1 caused an obvious fluorescence quenching, the introduction of other metal ions of Na+, K+, Ca2+, Mg2+, Ba2+, Pb2+, Al3+, Fe3+, Zn2+, Cd2+, Cu2+ and Mn2+ had negligible effect to fluorescence intensity, whereas some metal ions like Ni2+ and Cr3+ slightly weaken the fluorescence. In general, all these ions would not induce any remarkable disturbance for detecting Hg2+ in the fluorescence change at 526 nm. In order to further test the interference of other common cations in recognition of Hg2+, the competition experiments were performed: sensor 1 was conducted with the addition of 1.0 equiv. Hg2+ to induced fluorescence quenching before mixed 10 equiv. Na+, K+, Ca2+, Mg2+, Cd2+, Ba2+, Fe3+, Pb2+, Al3+, Ni2+, Cr3+, Mn2+ and Zn2+ respectively. The fluorescence intensity of the mixed system at 526 nm was shown in Fig. 6 (red bar), all of them exhibited still quenching, so the experimental results indicated that these ions had no interference for Hg2+ detection. Thus the fluorometric analysis above had proven that sensor 1 could serve as an outstanding sensitive and selective fluorescent sensor for Hg2+ in our prospective.
 |
| | Fig. 6 Fluorescence intensities of sensor 1 (1 μM L−1) in the presence of various metal ions (black bar) and competition experiment (red bar) in Tris–HCl solution [V(C2H5OH)–V(H2O) = 7:3, pH = 7.2] (λex = 368 nm, λem = 526 nm). | |
We selected the filter paper as the supporter of the sensor 1 to make a dipstick for the Hg2+ detecting. After dropping a solution of sensor 1 on the neutral filter paper and drying it, a fluorescent dipstick was formed. When exposed the modified filter-paper to the aqueous solution of various metal ions under UV lamp, only Hg2+ induced the fluorescence quenching as shown in Fig. 7.
 |
| | Fig. 7 Fluorescence changes of filter paper containing sensor 1 treated with various metal ions under UV lamp. | |
3.3 Detection of Hg2+ in living cells
To explore the potential biological application of this sensor, we researched the capability of sensor 1 to track Hg2+ in living GES-1 (human stomach lining cells) cell. The images were obtained upon irradiation at 405 nm with a band path from 395 to 425 nm under identical exposure conditions. The living cells were first incubated with 0.1 mmol L−1 sensor 1 in DMF for 30 min at 37 °C in 5% CO2 atmosphere, then washed the cells with phosphate buffered saline (PBS, pH = 7.4) 3 times, the strong intracellular fluorescence illustrate the sufficient accumulation of sensor 1 in the living cells (Fig. 8a). However, after adding 0.2 mmol L−1 Hg2+ in to the cells which supplemented with sensor 1, the intracellular fluorescence quenched as shown in Fig. 8b. So Fig. 8 displays the fluorescence distinction of sensor 1 before and after treated with Hg2+ in living GES-1 cells, illustrating that it can be a valuable molecular sensor for studying biological processes involving Hg2+ within living cells.
 |
| | Fig. 8 Fluorescence microscope imaging of GES-1 cells stained with 1 × 10−4 mol L−1 sensor 1 before and after treating with 2 × 10−4 mol L−1 of Hg2+. | |
4. Conclusions
We have developed a fluorescent chemosensor for recognizing Hg2+ by the conjugation of quinoline and phenothiazine. It displayed excellent selectivity and sensitivity to Hg2+ with a 1:1 binding mode. Upon addition common metal ions, only Hg2+ caused a striking fluorescence quenching. Moreover, the cell-permeable experiment indicated this sensor can indeed visualize the change of intracellular Hg2+ in living cells. We expect that such Hg2+ sensor will have broad application in biomedical and environmental detections.
References
-
(a) A. Jana, J. S. H. Kim, S. Jung and P. K. Bharadwaj, Chem. Commun., 2009, 4417 RSC;
(b) D. W. Boening, Chemosphere, 2000, 40, 1335 CrossRef CAS;
(c) H. H. Harris, I. J. Pickering and G. N. George, Struct. Sci., 2003, 301, 1203 CAS.
-
(a) M. Harada, Crit. Rev. Toxicol., 1995, 25, 1 CrossRef CAS PubMed;
(b) N. K. Mottet, M. E. Vahter, J. S. Charleston and L. T. Friberg, Met. Ions Biol. Syst., 1997, 34, 371 CAS;
(c) T. W. Clarkson, L. Magos and G. J. Myers, N. Engl. J. Med., 2003, 349, 1731 CrossRef CAS PubMed.
-
(a) Q. Wang, D. Kim, D. D. Dionysiou, G. A. Sorial and D. Timberlake, Environ. Pollut., 2004, 131, 323 CrossRef CAS PubMed;
(b) P. B. Tchounwou, W. K. Ayensu, N. Ninashvili and D. Sutton, Environ. Toxicol., 2003, 18, 149 CrossRef CAS PubMed;
(c) R. Eisler, Environ. Geochem. Health, 2003, 25, 325 CrossRef CAS.
-
(a) E. M. Nolan and S. J. Lippard, Chem. Rev., 2008, 108, 3443–3480 CrossRef CAS PubMed;
(b) Q. He, E. W. Miller, A. P. Wong and C. J. Chang, J. Am. Chem. Soc., 2006, 128, 9316–9317 CrossRef CAS PubMed;
(c) K. Harano, S. Hiraoka and M. Shionoya, J. Am. Chem. Soc., 2007, 129, 5300 CrossRef CAS PubMed;
(d) M. Leermakers, W. Baeyens, P. Quevauviller and M. Horvat, Trends Anal. Chem., 2005, 24, 383 CrossRef CAS PubMed.
-
(a) L. Zeng, E. W. Miller, A. Pralle, E. Y. Isacoff and C. J. Chang, J. Am. Chem. Soc., 2006, 128, 10 CrossRef CAS PubMed;
(b) G. Farruggia, S. Iotti, L. Prodi, N. Zaccheroni, M. Montalti, P. B. Savage, G. Andreani, V. Trapani and F. I. Wolf, J. Fluoresc., 2008, 19, 11 CrossRef PubMed.
-
(a) H. Komatsu, T. Miki, D. Citterio, T. Kubota, Y. Schindo, Y. Kitamura, K. Oka and K. Suzuki, J. Am. Chem. Soc., 2005, 127, 10798 CrossRef CAS PubMed;
(b) M. Taki, M. Desaki, A. Ojida, S. Iyoshi, T. Hirayama, I. Hamachi and Y. Yamamoto, J. Am. Chem. Soc., 2008, 130, 12564 CrossRef CAS PubMed;
(c) Q. He, E. W. Miller, A. P. Wong and C. J. Chang, J. Am. Chem. Soc., 2006, 128, 9316 CrossRef CAS PubMed;
(d) B. K. Kanungo, M. Baral, R. K. Bera and S. K. Sahoo, Monatsh. Chem., 2010, 141, 157 CrossRef CAS;
(e) R. Akbar, M. Baral and B. K. Kanungo, Spectrochim. Acta, Part A, 2014, 129, 365 CrossRef CAS PubMed.
- H. Yang, Z. Zho, K. Huang, M. Yu, F. Li, T. Yi and C. Huang, Org. Lett., 2007, 9, 4729 CrossRef CAS PubMed.
-
(a) H. S. Jung, P. S. Kwon, J. W. Lee, J. I. Kim, C. S. Hong, J. W. Kim, S. Yan, J. Y. Lee, J. H. Lee, T. Joo and J. S. Kim, J. Am. Chem. Soc., 2009, 131, 2008 CrossRef CAS PubMed;
(b) E. M. Nolan and S. J. Lippard, Acc. Chem. Res., 2009, 42, 193 CrossRef CAS PubMed.
- S. Goswami, K. Aich, A. K. Das, A. Manna and S. Das, RSC Adv., 2013, 3, 2412 RSC.
-
(a) H. A. Benesi and J. H. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703 CrossRef CAS;
(b) M. Barra, C. Bohne and J. C. Scaiano, J. Am. Chem. Soc., 1990, 112, 8075 CrossRef CAS;
(c) M. Zhu, M. J. Yuan, X. F. Liu, J. L. Xu, J. Lv, C. S. Huang, H. B. Liu, Y. L. Li, S. Wang and D. B. Zhu, Org. Lett., 2008, 10, 1481 CrossRef CAS PubMed.
- M. D. Sun, S. D. Wang, Q. B. Yang, X. L. Fei, Y. X. Li and Y. P. Li, RSC Adv., 2014, 4, 8295 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra03908d |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.