Efficient green synthesis of bis(cyclic carbonate)poly(dimethylsiloxane) derivative using CO2 addition: a novel precursor for synthesis of urethanes
K. R. Aguiara,
V. G. Santosb,
M. N. Eberlinb,
K. Rischkac,
M. Noeskec,
G. Tremiliosi-Filhoa and
U. P. Rodrigues-Filho*a
aUniversity of Sao Paulo, Institute of Chemistry of Sao Carlos, Sao Carlos, Brazil. E-mail: uprf@iqsc.usp.br
bState University of Campinas, ThoMSon Mass Spectrometry Laboratory, Campinas, Brazil
cFraunhofer-Institut für Fertigungstechnik und Angewandte Materialforschung IFAM, Bremen, Germany
First published on 23rd May 2014
Abstract
Poly(dimethyl siloxane), PDMS, with terminal cyclic carbonate groups was prepared by cycloaddition of carbon dioxide to epoxy rings using tetra alkyl-ammonium bromide as a catalyst under efficient and mild conditions. The process was carried out under a modest pressure of carbon dioxide (i.e., <2 MPa) and temperature (<200 °C). The oligomeric species was found to be a non-Newtonian fluid with low molecular weight (MW ∼ 1200 g mol−1) with n = xx − yy and thermally stable up to 200 °C. Its formation was verified by mid infrared spectroscopy (FT-MIR), Matrix-Assisted Laser Desorption Ionization Mass Spectroscopy MALDI-Tof-MS and multinuclear magnetic resonance (13C NMR, 29Si NMR, 1H NMR). The urethane synthesis was confirmed by FTIR, NMR and X-ray Photoelectron Spectroscopy (XPS) after reacting amines or diamines, in mild reactions conditions, with bis(cyclic carbonate)poly(dimethylsiloxane).
Introduction
Carbon dioxide (CO2) is the primary greenhouse gas emitted through human activities and believed to be partly responsible for a change of the global climate. CO2 is naturally present in the atmosphere as part of the Earth's carbon cycle, but human activities such as burning of fossil fuels, mineral extraction, electronic industries, steel mills, cement industries and bioethanol production contributes greatly by releasing carbon dioxide to the atmosphere. As CO2 is also regarded as a cheap and green carbon resource1,2 it becomes an attractive raw material for incorporation into industrial processes such as a monomers and solvents in the manufacturing of biodegradable copolymers, most notably, polycarbonates.3 CO2 can act as a building block for organic carbonates, supplanting phosgene, and for carboxylates, avoiding multistep procedures.4 Recently, the chemistry of CO2 and its fixation have received great deal of attention due to the utilization of the least-expensive carbon source, reducing global warming gas emission.5 One of the most promising methodologies in this area is the synthesis of cyclic carbonates via cycloaddition of CO2 into oxirane rings6 of organic epoxides. The reaction of carbon dioxide with oxiranes to produce cyclic carbonates has been considered a useful method for CO2 fixation by chemical processes. Cyclic carbonates has received remarkable attention as aprotic polar solvent, sources for polymer synthesis7–9 and as electrolytes for lithium ion batteries.10 Organic solvents with five-membered cyclic carbonates scarcely polymerize as a result of the stable five-membered rings,11 whereas they efficiently react with amines to afford the corresponding hydroxyurethanes,12 thus this chemical route is applicable for polyurethane synthesis. Therefore, the aim of this work is to present a Green route permitting an easy scale-up to obtain a bis(cyclic carbonate)-PDMS derivative by cycloaddition reaction of carbon dioxide and epoxide to afford the formation of a precursor for synthesis of urethanic-PDMS material under mild and metal-free conditions.Experimental
Materials
Poly(dimethylsiloxane) diglycidylether terminated (PDMS, 15 cSt at 25 °C, Mn ∼ 800 g mol−1, 0.99 g mL−1 at 25 °C), tetraethylammonium bromide (TEAB, 98%), 2-ethoxyethanol (EG, ≥99%), 3-aminomethyl-3,5,5-trimethylcyclohexylamine (IPDA, >98%, 0.94 g mL−1 at 20 °C), phenylethylamine (PEA, 99%, 0.99 g mL−1 at 25 °C), poly(bisphenol A carbonate) (PC) were supplied by Sigma Aldrich and used as received.The 1-methyl-2-pyrrolidone (NMP, 99%) was provided by Vetec and carbon dioxide (CO2, 99.99%) by Linde Gas.
Reactor design
Reactor Parr Model 2192HC4 was used in the synthesis of cyclic carbonate. The used cylinder volume was 100 mL, stainless steel T316SS, maximum temperature of 225 °C and maximum pressure of 13.1 MPa.Preparation of bis(cyclic carbonate)PDMS derivative – CCPDMS
The CCPDMS proposed in this work was prepared using suitable quantities of PDMS, catalyst and solvent, as shown in Table 1. After purging the cylinder with CO2, the PDMS and TEBA were transferred to the cylinder and dissolved in ethoxyethanol. In sequence, the carbon dioxide was charged in the reactor and the pressure was set to 0.551 MPa, 0.827 MPa or 1.10 MPa at 150 °C. The autoclave was heated at that temperature for 8 h, and the pressure was kept constant during the reaction. After reaction, the autoclave was allowed to cool down and then the excess of CO2 was released. The liquid was transferred to a Petri dish and the solvent was evaporated at 70 °C.| Reactants | Reaction conditions | ||
|---|---|---|---|
| PDMS | 9.94 g (12.42 mmol) | Temperature | 150 °C ± 1 |
| TEBA | 0.099 g (1% weight) | Pressure | 0.551; 0.827 or 1.10 MPa |
| EG | 40 mL | Time | 8 h |
Urethane synthesis from CCPDMS
The mass of 1 mmol of CCPDMS was weighed in a closed vessel, previously purged with nitrogen. 3 mmol of IPDA or PEA were added and the reaction took place for 40 to 100 minutes at 70 °C, without solvent. Before characterizations, the urethanes products resulting from CCPDMS, denoted as PDMSUr, were washed with deionized water at 70 °C for 20 minutes, in order to remove remaining TEBA. PDMSUr were also washed with HCl 1 M to remove unreacted amines.Characterizations
A BOMEM MB102 FTIR spectrometer was employed to analyse previously the formation of the cyclic carbonate group and urethanic bonds in the range 250–4000 cm−1, 32 scans and instrumental resolution set at 4 cm−1 in attenuated total reflectance (FTIR-ATR) or transmission (FTIR-T) mode of analysis were applied. The samples were cast on a silicon wafer before the measurements. To carry on the quantitative analysis of CCPDMS by FTIR, a 25 μm thick silicon wafer was used on transmission FTIR measurements. The thermogravimetric analysis (TGA) was performed in a 2950 TGA HR V5.4A (TA instruments) from 30 to 1000 °C at a heating rate of 10 K min−1, nitrogen flow rate of 50 mL min−1 and sample mass about 30 mg. 1H and 13C NMR spectra of CCPDMS were recorded in an Agilent 500 MHz with acquisition parameters as follows: (1H) scan numbers (NS) 16, pulse widths (P1) 3.95 μs, relaxation delay (D1) 1s; (13C) NS = 4096, P1 = 6.6 μs and D1 = 3 s. 1H and 13C NMR spectra of PDMS were recorded in an Agilent400 MHz with the following parameters: (1H) NS = 16, P1 = 4.45 μs; (13C) D1 = 1 s; NS = 10.000, P1 = 5.85 μs and D1 = 3 s. All spectra were referenced to CDCl3 at 7.26 ppm. The 29Si NMR spectrum was taken in a Varian Innova (29Si resonance frequency 79.46 MHz), the samples were dissolved in CDCl3 (with 99.96% of the hydrogen atoms being deuterium atoms), tetramethylsilane (TMS) was used as the internal standard (0 ppm), NS = 1989, P1 = 9.88 μs and D1 = 25 s.13C NMR spectra of urethanes were acquired in a Bruker spectrometer in CDCl3, (13C) NS = 34![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 816, P1 = 4 μs and D1 = 1 s. XPS spectra were taken using a Kratos Ultra system applying the following acquisition parameters: base pressure: 4.10−8 Pa, sample neutralisation applying low energy electrons (<5 eV), hybrid mode (electrostatic and magnetic lenses are used), take off angle of electrons 0°, pass energy 20 eV (or, respectively, 40 eV in case of some N1s spectra) in high resolution spectra and 160 eV in survey spectra, excitation of photoelectrons by monochromatic AlKα radiation. The analysis area is elliptically shaped with main axes of 300 μm × 700 μm. The binding energy calibration of the electrically isolating polymer was performed by referring the C1s component of aliphatic carbon species to 285.0 eV. Binding energies are given with a precision of ±0.1 eV throughout this contribution and peak widths are indicated as full width at half maximum (FWHM) with a precision of ±0.05 eV. Elemental ratio was calculated based on the area of the peaks and considering the percentage of the elements provided by XPS. Viscosity measurements were carried out in a cone-plate viscometer Brookfield CAP 2000, at 50 °C during 15 s for each shear rate: 6.66 × 102 s−1, 1.33 × 103 s−1, 2.66 × 103 s−1 and 5.33 × 103 s−1. The percentage of bromine was measured by X-ray fluorescence analysis (XRF) with a MiniPal 4-Panalytical, applying 9 W to a Rh tube, acquisition time was set to 15 minutes under He atmosphere. Elemental analysis (EA) was performed on a Perkin-Elmer CHN 2400. For MALDI-TOF an aliquot of 1 μL was spotted on the MALDI plate, allowed to dry and then covered with 1 μL of 9-amino matrix (9-aminocridine hemihydrate), 10 mg mL−1 in 60/40 (v/v) isopropanol–acetonitrile. The experiments were carried out by using a MALDI-TOF/TOF Autoflex III (Bruker Daltonics, Germany) equipped with a nitrogen laser irradiating at λ = 332 nm. Each spectrum was acquired from 500 laser shots on a single spot in the linear positive ion mode. The m/z range over which ions were detected was m/z 700 to 3000. Measurements were done in operating conditions as follows: ion source 1 = 19.00 kV, ion source 2 = 16.72 kV, lens voltage = 8.30 kV, reflector voltage 1 = 21.00 kV, reflector voltage 2 = 9.70 kV, pulsed ion extraction time = 40 ns, suppression = 500 Da.
816, P1 = 4 μs and D1 = 1 s. XPS spectra were taken using a Kratos Ultra system applying the following acquisition parameters: base pressure: 4.10−8 Pa, sample neutralisation applying low energy electrons (<5 eV), hybrid mode (electrostatic and magnetic lenses are used), take off angle of electrons 0°, pass energy 20 eV (or, respectively, 40 eV in case of some N1s spectra) in high resolution spectra and 160 eV in survey spectra, excitation of photoelectrons by monochromatic AlKα radiation. The analysis area is elliptically shaped with main axes of 300 μm × 700 μm. The binding energy calibration of the electrically isolating polymer was performed by referring the C1s component of aliphatic carbon species to 285.0 eV. Binding energies are given with a precision of ±0.1 eV throughout this contribution and peak widths are indicated as full width at half maximum (FWHM) with a precision of ±0.05 eV. Elemental ratio was calculated based on the area of the peaks and considering the percentage of the elements provided by XPS. Viscosity measurements were carried out in a cone-plate viscometer Brookfield CAP 2000, at 50 °C during 15 s for each shear rate: 6.66 × 102 s−1, 1.33 × 103 s−1, 2.66 × 103 s−1 and 5.33 × 103 s−1. The percentage of bromine was measured by X-ray fluorescence analysis (XRF) with a MiniPal 4-Panalytical, applying 9 W to a Rh tube, acquisition time was set to 15 minutes under He atmosphere. Elemental analysis (EA) was performed on a Perkin-Elmer CHN 2400. For MALDI-TOF an aliquot of 1 μL was spotted on the MALDI plate, allowed to dry and then covered with 1 μL of 9-amino matrix (9-aminocridine hemihydrate), 10 mg mL−1 in 60/40 (v/v) isopropanol–acetonitrile. The experiments were carried out by using a MALDI-TOF/TOF Autoflex III (Bruker Daltonics, Germany) equipped with a nitrogen laser irradiating at λ = 332 nm. Each spectrum was acquired from 500 laser shots on a single spot in the linear positive ion mode. The m/z range over which ions were detected was m/z 700 to 3000. Measurements were done in operating conditions as follows: ion source 1 = 19.00 kV, ion source 2 = 16.72 kV, lens voltage = 8.30 kV, reflector voltage 1 = 21.00 kV, reflector voltage 2 = 9.70 kV, pulsed ion extraction time = 40 ns, suppression = 500 Da.
Yield of the synthesis of CCPDMS
The reaction yield was calculated by mid-infrared spectroscopy using an analytical curve from infrared absorption of the carbonyl group (1774 cm−1)13 versus concentration of the PC standard solutions (3.0, 3.5, 4.0, 4.5 and 5.0% by weight) in NMP. Thus, by data interpolation employing the calibration curve equation y = 0.1012x − 0.1072, where x is the absorbance of carbonyl groups in FTIR and y the concentration in wt%, was possible to determine the carbonate concentration and the yield of the reaction.Results
Synthesis of CCPDMS
Scheme 1 shows the cycloaddition of CO2 to the oxirane ring and the expected formation of bis(cyclic carbonate). Quaternary ammonium salts are currently used as cheap and efficient homogeneous catalysts for the industrial preparation of cyclic carbonates. In 2002, Caló and co-workers reported cyclic carbonate synthesis from CO2 and oxiranes using the tetrabutylammonium halides as solvents and catalyst.14 Halide salts show high catalytic activity and the order of intrinsic activity follows: chloride > bromide > iodide, according to the nucleophilicity of the anion.15 The reaction is supposed to proceed via nucleophilic attack of halide to oxirane to form β-haloalkoxide which reacts with CO2 followed by cyclization. | ||
| Scheme 1 Reaction of CO2 with a diepoxide forming bis(cyclic carbonate)PDMS derivative. | ||
The reaction rate depends on the nucleophilicity of the halide ion as well as the structure of the cation.14,15 Steric hindrance created from rings is another factor which implies in selective CO2 fixation reaction. Cyclohexene oxide PDMS derivative, e.g., required a prolonged reaction time, without significant yields, according to experimental procedures performed in our group and literature reports.16,17 Scheme 2 shows the proposed mechanism.
 | ||
| Scheme 2 Mechanism for the catalysed cycloaddition reaction of carbon dioxide and oxiranes rings. | ||
The infrared spectroscopy (FTIR) previously confirms the conversion of epoxy rings into five-membered cyclic carbonate after reaction with carbon dioxide. The band corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of carbonyl group of cyclic carbonate occurs at 1800 cm−1.17,18 Fig. 1 shows the superimposed spectra of PDMS and CCPDMS. The spectrum presented in Fig. 1 shows a doublet at 1100 cm−1 and 1020 cm−1 that corresponds to asymmetric (νass) and symmetric (νs) stretching vibration, respectively, of the two neighbour siloxane bonds.19 The absorptions at 1259 cm−1 and 800 cm−1 are assigned to in-plane bending or scissoring and out-of-plane oscillations of the Si–CH3 bonding, respectively.20,21 Because of the high coefficient of absorption of siloxane bonds in the region 1000–1100 cm−1, the symmetric stretching of epoxy ring at 1250 cm−1 and asymmetric ring stretching band of epoxy ring at 910 cm−1 are not visible in the spectrum. Therefore, to visualize the changes in the epoxy band, a curve fitting was performed using Winspec software. Fig. 2 shows the Gaussian functions simulating the experimental peaks of FTIR/T and the residue of CCPDMS and PDMS.
O stretching of carbonyl group of cyclic carbonate occurs at 1800 cm−1.17,18 Fig. 1 shows the superimposed spectra of PDMS and CCPDMS. The spectrum presented in Fig. 1 shows a doublet at 1100 cm−1 and 1020 cm−1 that corresponds to asymmetric (νass) and symmetric (νs) stretching vibration, respectively, of the two neighbour siloxane bonds.19 The absorptions at 1259 cm−1 and 800 cm−1 are assigned to in-plane bending or scissoring and out-of-plane oscillations of the Si–CH3 bonding, respectively.20,21 Because of the high coefficient of absorption of siloxane bonds in the region 1000–1100 cm−1, the symmetric stretching of epoxy ring at 1250 cm−1 and asymmetric ring stretching band of epoxy ring at 910 cm−1 are not visible in the spectrum. Therefore, to visualize the changes in the epoxy band, a curve fitting was performed using Winspec software. Fig. 2 shows the Gaussian functions simulating the experimental peaks of FTIR/T and the residue of CCPDMS and PDMS.
 | ||
| Fig. 1 Superimposed FTIR-T spectra of PDMS (gray) and CCPDMS (black). | ||
 | ||
| Fig. 2 Gaussian components obtained from the adjustment of FTIR spectra of PDMS and CCPDMS in the region 1100–800 cm−1. The fitting residue is shown below each spectrum. | ||
The best curve fitting of CCPDMS and PDMS in the region 1100–800 cm−1 was reached with five main Gaussian functions which centroids are nearby 1100, 1023, 910, 820 and 800 cm−1. This fitting revealed the peak of epoxy groups in PDMS (assigned by a narrow) and its disappearance after cycloaddition.
The structure of the CCPDMS was evaluated by 13C, 1H and 29Si nuclear magnetic resonance. Table 2 shows the carbon numbering in the PDMS and CCPDMS. Fig. 3 shows 13C NMR spectra of the acquired PDMS and the synthesized CCPDMS materials. The cycloaddition of CO2 in the epoxy ring is confirmed and resulted in characteristics signals of cyclic carbonate at δ 154.8 ppm (attributed to carbon 1, CO),22 66.3 ppm (carbon 2, –CH2–O–) and 74.7 ppm (–CH–, carbon 3).22–24 The peaks detected between 69–75 ppm are attributed to carbons of ether linkage (carbons 4 and 5).22–24 Signals assigned to carbons 6 and 7 appear at δ 23.3 and 14 ppm, respectively, and peaks resulting from carbons 8 and 9 are observed between 2 and 0.54.21,24 The signals corresponding to carbon atoms in epoxy groups are observed around 44 and 50 ppm.22
| Chemical structure | Code |
|---|---|
 |
CCPDMS |
 |
PDMS |
 | ||
| Fig. 3 13C NMR spectra of PDMS and CCPDMS in CDCl3. | ||
Comparing the spectrum obtained for CCPDMS with the one measured for PDMS, the signal at 44.3 ppm still remains, but is less intense than observed for PDMS due to the conversion of epoxy into five membered cyclic groups. The insertion of CO2 into oxiranes ring to form the bis(cyclic carbonate) functionality is efficient and high yield is obtained under mild conditions.
Fig. 4 shows the 1H NMR spectrum of CCPDMS and the characteristics signals of protons at the carbonate ring as well the peaks of protons at unreacted epoxy groups, assigned by the arrows. The peaks between 0 and 1.6 ppm are attributed to the protons CH2 and –Si–(CH3)2– (hydrogens atoms 6, 7, 8 and 9) from the poly(dimethylsiloxane) chain. The hydrogens atoms of the ether functional group (hydrogen 4 and 5) correspond to signals measured at about 3.3 ppm to 3.6 ppm, and the characteristics protons of the cyclic carbonate (2 and 3) bring about the peaks in the range of 4.2 and 4.8 ppm.23,25 The remaining protons from epoxy groups are visualized in the spectra assigned by the arrows for chemical shift values between 3.0 and 2.6 ppm.22
 | ||
| Fig. 4 1H NMR spectra of CCPDMS synthesized by fixation of carbon dioxide. | ||
Less than 1% of epoxy groups remained without conversion into bis(cyclic carbonate). Fig. 5 displays the 29Si NMR spectra of CCPDMS with peaks at 7.76 ppm and −21.79 ppm. The latter peak corresponds to a D2 environment: [–Si(CH3)2–]n or PDMS.21,26 The PDMS backbone and side groups remain unchanged after the reaction of the terminal oxiranes groups with carbon dioxide. Summarizing, the results observed by the FTIR and NMR revealed the formation of the proposed bifunctional five membered cyclic carbonate PDMS derivative.
 | ||
| Fig. 5 29Si NMR 400 MHz spectra of CCPDMS (CDCl3). | ||
In the XPS spectrum of the CCPDMS, peaks corresponding to carbon, oxygen and silicon atoms were detected. The clearly observed Si (2p) signal at a binding energy of 102.3 eV is attributed to silicone.27 The O1s signal was fitted with three Gaussian–Lorentzian peaks at 533.6 eV, 532.4 eV, 534.6 eV, which correspond to oxygen atoms O* bonded ether like C–O*–C, CO*/Si–O* and –O*–CO–O*, respectively.28–31 Based on the areas obtained after fitting the O 1s region, the ratio of atomic O* concentrations [–O*–CO–O*–]/[C–O*–C], was found to be 1.9.
This experimental ratio agrees with theoretical expectations of two oxygens atoms from cyclic carbonate for one ether linkage (2:1) in the spacer between the cyclic carbonate and the PDMS moieties, according to the proposed chemical structure. Fig. 6 shows as well the fitted spectra in the O1s and C1s regions as a table with theoretical and experimental atomic concentrations of the elements.
 | ||
| Fig. 6 XPS spectra of CCPDMS fitted with Gaussian–Lorentzian components in the O1s and C1s regions. | ||
The C1s region showed several contributions, with three of them being clearly perceivable at binding energies of 291.3 eV, approximately 287.6 eV, and 285 eV. Based on the proposed chemical structure, these peaks are interpreted to correspond to carbonate carbon atoms O–C*O–O (labelled C1 in the molecular representation of CCPDMS in Table 2), carbon atoms O–C*–C*–O in the carbonate ring (labelled C2 and C3), and aliphatic carbons C–C*/C*–H.28,29 Notably the carbonate signal is clearly identified due to its unique C1s binding energy of 291.3 eV,32 obtained for a signal with a width of 0.95 eV here. Moreover, the C1s signal contribution with a binding energy of 287.6 eV and a width of 1.15 eV is attributed to sp3 hybridized carbon atoms C*–O bonded to oxygen. The particularly high binding energy shift of +2.6 eV of such cyclic carbonate related C*O with respect to aliphatic carbon atoms is significantly higher than the shift reported for aromatic polycarbonates.28 It is related to the lack of π-electron conjugation in the neighbourhood of the carbonate group within the five-membered ring. Furthermore, signal contributions resulting from the ether carbon atoms C*O (labelled C5) and the ether carbon atoms close to the carbonate ring (labelled C4) are fitted with equal intensities. In this way, binding energies of 287.6 eV for C2 and C3, 286.9 eV for C4, and 286.1 eV for C5 are suggested, as displayed in Fig. 6. The remarkably high C1s binding energy for C4 is tentatively attributed to a secondary shift exercised by the carbon atoms of type C3. Based on the performed fitting, the respective atomic carbon concentration ratio [O–C*O–O] :[O–C*–C*–O] is found to amount to 2.6, which is in fair accordance with the ratio of 2 calculated using the proposed chemical structure. The atomic concentration ratio of [Si] :[C*O] was obtained from fitting the C1s signal region and considering the total carbon atomic concentration 57.3 at% obtained. Comparing the thus determined experimental ratio of 6.1 and the stoichiometric ratio of 6 based on the chemical formula and the FTIR and NMR investigations of the CCPDMS material, a sound concordance was found. It means that in case of n = 10, for each PDMS segment on the chain there are 2 carbonate groups. This is in good agreement with the expected chemical structure. XPS was a suitable technique to give information about the whole structure of the synthesized compound CCPDMS. Based on the bond lengths values was possible to estimate the size of the CCPDMS chain, which was around 4.5 nm. The information depth reached by XPS is approximately 10 nm is quite higher, thus information about the complete molecule is acquired by XPS analysis.33
Molecular mass and yield of CCPDMS
The CCPDMS produced after 8 h at 1.10 MPa of CO2 pressure and 150 °C was used to the calculate molecular mass by 1H NMR and oligomer distribution by MALDI-TOF-MS. The average molecular weight (Mn) was determined by 1H NMR end group analysis. Integrating the signals of proton on the end-groups and protons on the polymer chain (PDMS), simple math can be applied to deduce the Mn values and the average number n of dimethylsiloxane repetition units. The signal of end-group protons (hydrogen atoms 2 and 3) and protons of repeating monomer (8) of CCPDMS were integrated and from this value the yield was calculated. Table 3 shows Mn found to CCPDMS by 1H NMR and percentage of conversion into cyclic carbonate group.| % Conversion | |||
|---|---|---|---|
| 13C | 1H | FTIR | |
| CCPDMS | 93 | 85.6 | 94.1 |
Table 4 presents the elemental analysis of the CCPDMS. The findings confirm the carbon and hydrogen concentrations of the material expected on the base of stoichiometry calculated using the obtained n = 10. However, using XRF up to 3.6 mol% of bromine species were detected which are not a stoichiometric components of CCPDMS molecules. In an attempt to remove the bromide-containing catalyst TEBA, three washing procedures were performed in CCPDMS material. The samples before and after washing were analysed by XRF.
| % (mol) before washing | % (mol) after washing | |
|---|---|---|
| Bromineb | 3.6 | 0.96 |
In order to determine the full distribution of oligomers of CCPDMS, MALDI-TOF-MS (Fig. 7) was used. In the MALDI-TOF-MS of Fig. 7, a Gaussian distribution for protonated CCPDMS oligomers in the m/z 600–2800 range can be noticed which corresponds to an oligomeric distribution with n varying between 3 and 30 centered at around n = 11. The characteristic m/z separation of 74 units is consistent with increments of dimethylsiloxane units [–O–Si(CH3)2] comprising the most probable isotopes of O, Si, C and H and amounting to a mass of 74 Da.34 Also characteristic is the change in the isotopologue pattern of the ions (not shown) due to the increasing increments of silicon [28Si(100%), 29Si(5.06%) and 30Si(3.36%)] atoms. This characteristic and detailed MALDI-TOF-MS profile allows us to determine the oligomeric distribution of CCPDMS.
 | ||
| Fig. 7 MALDI mass spectrum of bis(cyclic carbonate)PDMS derivative CCPDMS. | ||
Three different CO2 pressures were tested: 0.551 MPa; 0.827 MPa and 1.10 MPa. The others variables were kept constant. The yield of cyclic carbonate at each pressure and time was determined by FTIR, based on the infrared absorption. The conversion of epoxy groups into carbonate groups is affected by the CO2 pressure. The yields by the time and carbon dioxide pressure are shown in Fig. 8.
 | ||
| Fig. 8 Yields of CCPDMS as a function of pressure and time during carbonate synthesis. | ||
Fig. 9 displays the TG and DTG curves, respectively, for CCPDMS obtained at a heating rate of 10 K min−1 under nitrogen. The main CCPDMS degradation is observed in the temperature range between 150 °C and 450 °C which coincides with observation reported by ref. 26. At around 200 °C a small weight loss (∼2.3%) is observed which probably occurs due the release of water, solvent, residuals monomers and others trapped impurities.33,36 Further increasing the temperature the main degradation stage occurs at 370 °C. This decomposition is attributed to evolution of methane and other hydrocarbonaceous gases from resulting homolytic scission of Si–CH3 bond33 and around 0.47 wt% of a silica residue was formed up to 650 °C.
 | ||
| Fig. 9 Thermogram of CCPDMS in N2 atmosphere. | ||
The dynamic viscosity of CCPDMS was evaluated (Fig. 10). Basically, the measurement and characterization of viscous behaviour of a fluid serve to multiple purposes such as determining the response of the fluid to deformations; it is an essential knowledge in developing critical specifications for handling, transportation, application, and in determining the optimum conditions for optimization of processing conditions. Moreover, understanding viscous behaviour contributes to the effective end use performance. From the plot in Fig. 10 it is noted that the viscosity decreases with shear rate. Such non-Newtonian behaviour indicates shear-thinning and is attributed in the molecular scale to a chain alignment with the flow direction. The polymer material orients in the flow direction thus showing a pseudoplastic characteristic.35 When CCPDMS is deformed there will be some slippage of chains over each other and molecular alignment in the direction of the applied stress.
 | ||
| Fig. 10 Rheological characterisation of CCPDMS with a plot of viscosity versus shear rate. | ||
Urethane synthesis from CCPDMS precursor
As carbonate precursors are reactive with respect to amines.37 CCPDMS may be expected to be applied as a precursor for low temperature and environmentally friendly route to urethanic materials. In order to test the reactivity of the synthesized CCPDMS material, a cyclic aliphatic diamine IPDA and an aromatic amine PEA were used. Based on infrared spectroscopy (FTIR) measurements, two new features of the resulting material were noticed when compared with CCPDMS spectrum, as shown in Fig. 11, displaying normalized spectra. Bands observed at 3200–3500 cm−1 ref. 21 and 35 are concluded to result from urethane bonds. Moreover, while the FTIR spectrum of CCPDMS shows an intense band at around 1800 cm−1, such signal corresponds to the stretching of the carbonyl band (νCO).38 After the reactions between CCPDMS and amines, that signal cannot be detected. For all spectra there is a doublet between 1100 cm−1 and 1020 cm−1, due to asymmetric (νass) and symmetric (νs) stretching of two neighboring siloxane bonds, respectively.19 Other characteristics bands at 1260 cm−1 and 800 cm−1 are attributed to axial deformation in plane and out-of-plane of the Si–CH3 bond (δSi–CH3 and ρSi–CH3, respectively).20,21 Bands corresponding to urethane bonds are observed at around 1700 cm−1 and 1534 cm−1 ref. 41 and 42 (dashed lines). It is concluded that a polyaddition reaction between CCPDMS and the added amines occurred, leading to the formation of a new urethanic material. The reaction occurred without any solvent and both products labelled PDMSUr–IPDA and PDMSUr–PEA formed gels (Fig. 11).
 | ||
| Fig. 11 FTIR-ATR superimposed spectra of CCPDMS and the urethanes produced with IPDA (labelled PDMSur–IPDA) and PEA (labelled PDMSUr–PEA). | ||
Additionally, 13C NMR and XPS investigations of PDMSUr–IPDA and PDMSUr–PEA were done in order to corroborate with FTIR results and to obtain more insight in chain segment length and the stoichiometry of the urethane materials. The corresponding 13C NMR spectra and general chemical structure are presented in Fig. 12. In both spectra, signals for carbon atoms at 14 ppm and 23 ppm are observed; they correspond to aliphatic and carbons from PDMS segments and appear between 2 and 0.54.19,23 The spectrum of PDMSUr–PEA resulting from the reaction with the aromatic diamine shows signals of aromatic carbons, and it also features a peak at around 156 ppm43,44 attributed to the characteristic carbon (CO, carbon 1) from urethane bond. The chemical shift from C-α (adjacent to the nitrogen, carbon 2) was inferred from the signal at 35 ppm, and C-β (carbon 3) originates a peak at 42 ppm.42 The reaction product of CCPDMS with the cycloaliphatic diamine PDMSUr–IDA presents major peaks at chemical shift values of 27.70, 31.74, 34.85 (C-α adjacent to the nitrogen, carbon 2), 36.13, 43.9, 46.7, 49.5, 54.83, 56.97, 66.32, 68.83, 71.33 and 74.14 ppm. Finally the peaks detected at 154.7, 156.6 and 156.9 ppm are attributed to N–13CO–O bonded carbon atoms in urethane bonds.41,44 No signal of carbonate like carbon atoms were observed in the spectra of PDMSUr–IDA and PDMSUr–PEA which indicates that a complete conversion of the carbonate groups in the CCPDMS precursor took place.
 | ||
| Fig. 12 13C NMR of PDMSUr–IDA and PDMSUr–PEA in CDCl3. | ||
Fig. 13 shows typical C1s and N1s spectra for PDMSUr–IPDA and PDMSUr–PEA after washing the substances with 1 M hydrochloric acid in order to remove possible unreacted amines. For both materials, the C1s core level spectra present three clearly distinguishable peak groups: a first peak at 285 eV attributed to hydrocarbonaceous carbon species (C*–C/, C*–H), a second peak at 286.6 eV comprising the signals of amine like (–C*–N), alcoholic (–C*–OH) and ether (–C*–O–C)28–30 species, and a third peak centered at 289.8 eV interpreted to result from a carbon species in a urethane (–NH–C*–O–O–) group.29,45
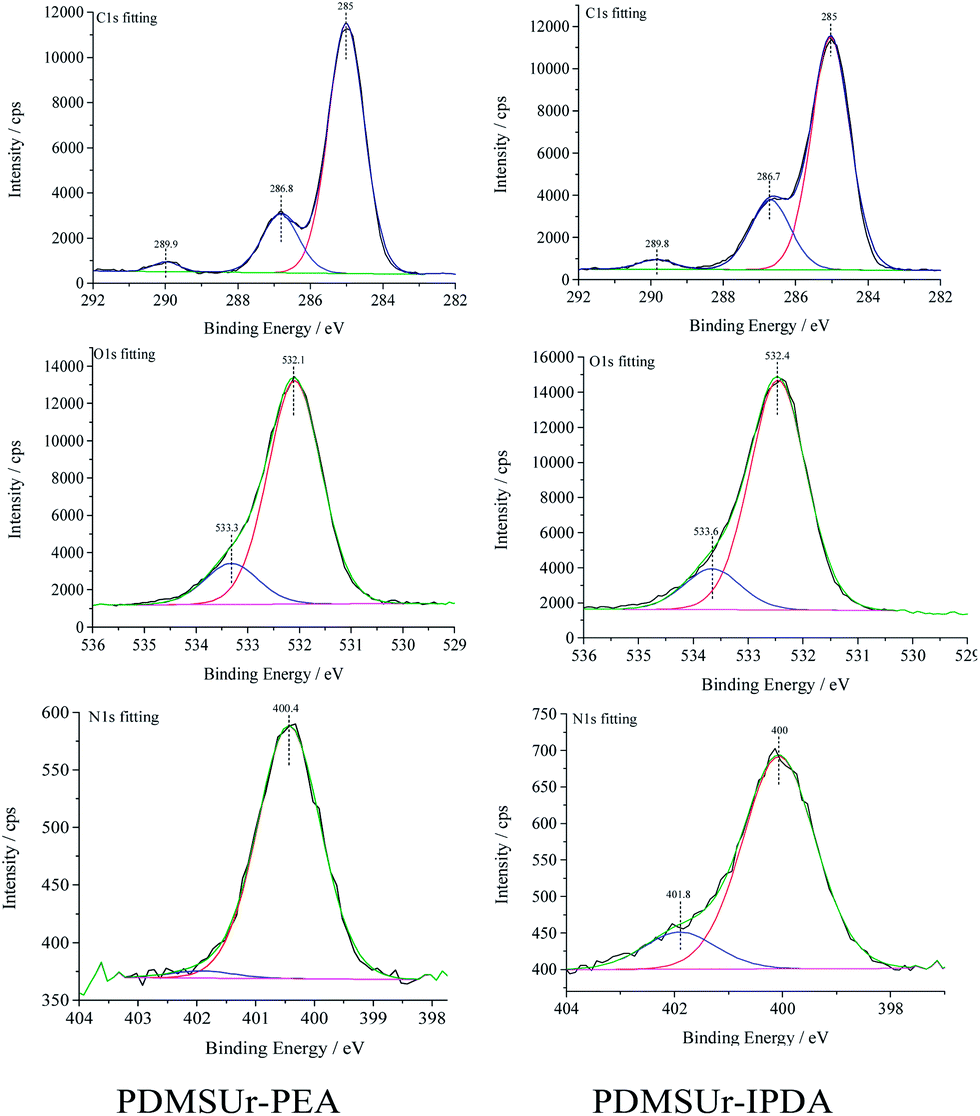 | ||
| Fig. 13 Fitting of XPS spectra obtained for PDMSUr–PEA and PDMSUr–IPDA in the C1s, O1s and N1s region. | ||
The N1s signal of PDMSUr–IPDA showed a shoulder at the higher binding energy (401.8 eV) side of the main signal at 400 eV and consequently was fitted with two peaks, whereas for PDMSUr–PEA a single peak at 400.4 eV gave a good match with the measured signal, as shown in the Fig. 13. For the PDMSUr–PEA, up to 97% of the nitrogen are assigned to urethane groups (–N*H–CO–O–)45 while PDMSUr–IPDA presented a contribution up to 85% of urethanic nitrogen. Nitrogen species with a binding energy of 401.5 eV contribute 15% of the total nitrogen in the sample;46,47 which correspond to 0.39 at% of N. Attributing the higher binding energy nitrogen feature at around 401.8 eV observed in the spectrum of PDMSUr–IDA to ammonium species gives rise to identify ca. 0.35% of ammonium hydrochloride48 species, which could be result as a product of the reaction between HCl hydrochloric acid and remaining free amines during the washing procedure. The ratio of nitrogen to urethane ([N]/[C*O]) and silicon to urethane ([Si]/[C*O]) was calculated based on the intensities of the signal identified as described above. The values are shown in Table 5. The N1s signal of PDMSUr–IPDA showed a shoulder at the higher binding energy (401.8 eV) side of the main signal at 400 eV and consequently was fitted with two peaks, whereas for PDMSUr–PEA a single peak at 400.4 eV gave a good match with the measured signal, as shown in the Fig. 13.
O–O and Si/CO ratio
| Code | %at C | %at N | %at O | %at Si | ||||
|---|---|---|---|---|---|---|---|---|
| The. | Exp. | The. | Exp. | The. | Exp. | The. | Exp. | |
| PDMSUr–PEA | 62.9 | 62.5 | 2.2 | 2 | 21.3 | 21.4 | 13.4 | 13.9 |
| PDMSUr–IPDA | 58.3 | 63 | 4.7 | 3 | 22.6 | 21 | 14.3 | 13 |
| [Si]/[N–C*O–O] |
[N]/[N–C*O–O] |
|||
|---|---|---|---|---|
| The. | Exp. | The. | Exp. | |
| PDMSUr–PEA | 6 | 6.6 | 1 | 1.3 |
| PDMSUr–IPDA | 6 | 7 | 1 | 1.3 |
The atomic concentration ratio [Si]/[N*–CO–O] for the PDMSUr–PEA is found to be in good agreement with the expected chemical structures, and experimental values concerning the composition of the material. The ratio between the total nitrogen content and the concentration of urethane groups, i.e. [N]/[N–C*O–O] gives an indication for the average molecular size of the urethane moieties synthesised. Regarding the atomic composition of PDMSUr–IPDA, the theoretical values were determined considering a polymeric structure (IPDA–CCPDMS)m–IPDA because IPDA like CCPDMS is a bi-functional compound and after the formation of the dimers IPDA–CCPDMS the addition reaction with further CCPDMS or IPDA moieties is expected to continue, and correspondingly, the chain length may be expected to continue increasing. The XPS results for PDMSUr–IPDA indicate that at least three PDMS segments are expected to be part of the chain and that the average oligomer is a heptamer, since the experimental ratio [N]/[N–C*O–O] of 1.3 is close to the one expected for m = 3. Therefore, the XPS results indicate that not only trimers IPDA–CCPDMS–IPDA (m = 1) with a resulting ratio [N]/[N–C*O–O] = 2 are formed. Table 6 shows the theoretical ratios for [N]/[N–CO–O].
O–O] for PDMSUr–IPDA with the composition (IPDA–CCPDMS)m–IPDA
| m | [N]/[N–C*O–O] |
Theoretical value |
|---|---|---|
| 1 | 4/2 | 2.0 |
| 2 | 6/4 | 1.5 |
| 3 | 8/6 | 1.3 |
| 4 | 1/8 | 1.2 |
Conclusions
We have successfully demonstrated the cycloaddition of CO2 to PDMS bis(glycidyl ether) terminated producing CCPDMS as a valuable and stable intermediate for terminal modification of PDMS. The applied strategies of synthesis use metal-free route mild conditions, and the synthesis results in a high yield. The relatively low viscosity of the bis(cyclic carbonate)PDMS allows the future use of this compound for solvent-free reactions with components containing nucleophilic groups like for instance amines, thiols or alkoxides. Reactions between CCPDMS and amines producing urethanes were shown to be feasible without using any solvent. The oligo-urethanes and poly-urethanes resulting from the reaction between primary amino groups of diamines and cyclic carbonates are expected to be linear molecules (with thermoplastic properties). However, reactions with cyclic carbonates and mixtures of diamines and triamines will result in crosslinked networks.Acknowledgements
The authors acknowledge the financial support by Fapesp within Grants 2011/06019-0,2011/08120-0 and 2013/05279-3; CNPQ and German academic exchange service (DAAD) from resources of the German Federal Ministry of Education and Research (BMBF) within the PROBAL-CAPES/DAAD project ID 57060300. The authors also thank Santa Catarina State University by viscometric measurements, Brazilian Agricultural Research Corporation (EMBRAPA) 29Si NMR analysis and University of Bremen, Germany for the 13C NMR.References
- D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058 CrossRef.
- D. S. Schime, Nature, 2011, 414, 169 CrossRef.
- D. J. Darensbourg, Chem. Rev., 2007, 107, 2388 CrossRef CAS.
- M. Aresta, Stud. Surf. Sci. Catal., 1998, 114, 65 CrossRef CAS.
- S.-E. Park, J.-S. Chang and K.-W. Lee, Stud. Surf. Sci. Catal., 2004, 153, 267 CrossRef.
- D. J. Darensbourg and M. W. Holtcamp, Coord. Chem. Rev., 1996, 153, 155 CrossRef CAS.
- R. Nomura, M. Kori and H. Matsuda, Makromol. Chem., Rapid Commun., 1988, 9, 739 CrossRef CAS.
- G. Rokicki, Makromol. Chem., 1985, 186, 331 CrossRef CAS.
- G. Rokicki and P. Jezewski, Polym. J., 1988, 20, 499 CrossRef CAS.
- B. Schäffner, et al., Chem. Rev., 2010, 110, 4554 CrossRef.
- L. Vogdanis, et al., Makromol. Chem., 1990, 191, 465 CrossRef CAS.
- T. Bürgel and M. Fedtke, Polym. Bull., 1993, 30, 61 CrossRef.
- B. A. Sweileh, Y. M. Al-Hiari, M. H. Kailani and H. A. Mohammad, Molecules, 2010, 15, 3661 CrossRef CAS.
- V. Caló, Org. Lett., 2002, 4, 2561 CrossRef.
- N. Kihara, N. Hara and T. Endo, J. Org. Chem., 1993, 58, 6198 CrossRef CAS.
- R. A. Watile, K. M. Deshmukh, K. P. Dhake and B. M. Bhanage, Catal. Sci. Technol., 2012, 2, 1051 CAS.
- L. Han, H.-J. Choi, S.-J. Choi, B. Lui and D.-W. Park, Green Chem., 2011, 13, 1023 RSC.
- E. Pretsch, P. Bühlmann and C. Affolter, Structure determination of organic compounds: tables of spectral data, Springer, 3rd. s.l, 2000 Search PubMed.
- J. N. Chazalviel and U. P. Rodrigues Filho, Thin Solid Films, 2012, 520, 3918 CrossRef CAS.
- F. Wang, Faculty of the Virginia Polytechnic Institute, s.n., Blacksburg, Virgínia, 1998 Search PubMed.
- L. F. Wang, Polymer, 2000, 41, 5083 CrossRef CAS.
- G. Ribeiro, et al., Tetrahedron Lett., 2008, 49, 6879 CrossRef CAS; D. Hoebbel, M. Nacker and H. Schmidt, J. Sol-Gel Sci. Technol., 1998, 12, 169 CrossRef.
- A.-L. Brocas, et al., J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2677 CrossRef CAS.
- Z. Zhu, et al., Macromolecules, 1994, 27, 4016 CrossRef.
- F. Joki-Korpela and T. T. Pakkanen, Eur. Polym. J., 2011, 47, 1694 CrossRef CAS.
- O. Foussaier, et al., Mater. Lett., 2000, 42, 305 CrossRef CAS.
- C. Perruchot, et al., Eur. Polym. J., 2004, 40, 2129 CrossRef CAS.
- G. Beamson and D. Brigs, High resolution XPS of organic polymers, John Willey and Sons, 1992 Search PubMed.
- D. P. Queiroz, A. M. Botelho do Rego and M. N. de Pinho, J. Membr. Sci., 2006, 281, 239 CrossRef CAS.
- H. Zhang, et al., BioResources, 2014, 9, 673 CAS.
- A. Qureshi1, et al., J. Phys.: Conf. Ser., 2010, 208, 1 Search PubMed.
- S. Nadimpali, J. Power Sources, 2012, 215, 145 CrossRef CAS; C. Harms, M. Wilhelm and G. Grathwohl, J. Membr. Sci., 2011, 383, 135 CrossRef.
- S. L. McArthur, G. Mishra and C. D. Easton, Applications of XPS in Biology and Biointerface Analysis, Springer International Publishing, 2014, ch. 2 CrossRef CAS; S. S. Pathak, A. Sharma and A. S. Khanna, Prog. Org. Coat., 2009, 65, 206 CrossRef CAS.
- G. Montaudo, M. S. Montaudo, C. Puglisis and F. Samperi, Rapid Commun. Mass Spectrom., 1995, 9, 1158 CrossRef CAS.
- R. N. Jana and G. B. Nando, J. Elastomers Plast., 2005, 37, 149 CrossRef CAS.
- S. K. Shukla, et al., Thermochim. Acta, 2004, 424, 209 CrossRef CAS.
- J. H. Clements, Ind. Eng. Chem. Res., 2003, 42, 663 CrossRef CAS.
- A. Stanciu, A. Airinei, D. Timpu, A. Ioanid, C. Ioan and V. Bulacovschi, Eur. Polym. J., 1999, 35, 1959 CrossRef CAS.
- E. Pretsch, P. Bühlmann and M. Badertscher, Structure determination of organic compounds, Springer, 4th edn, 2008, p. 14 Search PubMed.
- G. G. Suchkova and L. I. Maklakov, Vib. Spectrosc., 2009, 51, 333 CrossRef CAS.
- N. Bassam, C. Laure, B. Jean-Francois, R. Yann and M. Zephirin, Green Chem., 2013, 15, 1900 RSC.
- L. B. Corinne, J. C. David and B. Philip Shehan, Magn. Reson. Chem., 1993, 31, 222 CrossRef.
- L. Kun and E. M. Mark, J. Mater. Chem., 2012, 22, 18784 RSC.
- W. M. Richard and A. Taylor, Green Chem., 2004, 6, 151 RSC.
- E. Njungabe et al., Congrés Francais de Mécanique, 2013, http://hdl.handle.net/2042/52221.
- R. A. Andoa, G. M. do Nascimento, R. Landers and P. S. Santos, Spectrochim. Acta, Part A, 2008, 69, 319 CrossRef.
- A. J. Beck, et al., Plasma Processes Polym., 2005, 2, 64 CrossRef.
- M. Datta, H. J. Mathieu and D. Landolt, Appl. Surf. Sci., 1984, 18, 299 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |