
Effect of Interface energy and electron transfer on shape, plasmon resonance and SERS activity of supported surfactant-free gold nanoparticles
Abstract
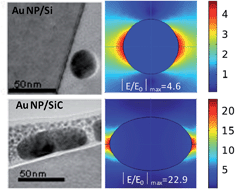
For device integration purposes plasmonic metal nanoparticles must be supported/deposited on substrates. Therefore, it is important to understand the interaction between surfactant-free plasmonic metal nanoparticles and different substrates, as well as to identify factors that drive nanoparticles nucleation and formation. Here we show that for nanoparticles grown directly on supports, the substrate/nanoparticle interfacial energy affects the equilibrium shape of nanoparticles. Therefore, oblate, spherical and prolate Au nanoparticles (NPs) with different shapes have been deposited by radiofrequency sputtering on substrates with different characteristics, namely a dielectric oxide Al2O3 (0001), a narrow bandgap semiconductor Si (100), and a polar piezoelectric wide bandgap semiconductor 4H–SiC (0001). We demonstrate that the higher the substrate surface energy, the higher the interaction with the substrate, resulting in flat prolate Au nanoparticles. The resulting localized surface plasmon resonance characteristics of Au NPs/Al2O3, Au NPs/Si and Au NPs/SiC have been determined by spectroscopic ellipsometry and correlated with their structure and shape studied by transmission electron microscopy. Finally, we have demonstrated the diverse response of the tailored plasmonic substrates as ultrasensitive SERS chemical sensors. Flat oblates Au NPs on SiC result in an enhanced and more stable SERS response. The experimental findings are validated by numerical simulations of electromagnetic fields.
 Please wait while we load your content...
Please wait while we load your content...

